



Guide to Understanding PDB data
Introduction to PDB data
•
PDB data
Biological molecule의 여러 meta data (coordinate 등)가 저장된 data format 을 의미한다.
널리 사용되는 file format: PDB, mmCIF, XML
각 file은 일반적으로 아래 세 가지 요소로 구성되어 있다.
◦
header section: protein에 대한 개략적인 정보, 어떤 방법으로 구조 얻었는지 등
◦
sequence: 서열 정보
◦
atom & coordinate: 각 atom의 이름, 특성, 좌표 정보 등
•
Visualization
참고) RCSB에서 default로 제공하는 시각화 tool은 Mol* viewer 이다.
•
Reading Coordinate Files
주로 chain ID와 atom 정보를 통해 원하는 부분의 좌표를 알아낼 수 있다.
Crystallography 구조의 경우, atom들에 temperature factor 등의 주석이 달려 있다.
NMR 구조의 경우, molecule의 여러 model을 포함하고 있는 경우가 일반적이다.
•
Potential Challenges
전세계의 연구자들이 가공해서 올리다보니, 특정 부분이 누락되어 있거나 (실험에서 관측이 안 되어서), 여러 atom type 중 position만 포함되어 있거나, loop이 없거나, 특정 domain 전체의 좌표/서열이 없는 등 다양한 이슈가 존재한다.
참고) Crystallography 구조의 경우 대부분 hydrogen atom 정보가 없다.
Beginner’s Guide to PDB structures
Traditional PDB file format (.pdb)
•
PDB file format (.pdb)
전통적으로 널리 사용되어 왔던 파일 형식이다.
하지만 점차 더 많은 chain 수, 더 다양한 molecule type 등을 하나의 파일에 효율적이고 분명하게 저장하기 위해 최근에는 PDBx/mmCIF file format (.cif) 사용이 권장된다.
•
PDBx/mmCIF file format (.cif)
현재 (@5/27/2025) 기준 최신 데이터 형식이다.
참고) 초기 CIF (Crystallographic Information File) 형식은 small molecule을 archive하기 위해 만들어졌고, 이후에 small molecule 이외에도 macromolecular crystallography 정보를 담기 위해 mmCIF 형식으로 확장되었다.
Dealing with Coordinates
RCSB PDB archive에 저장된 coordinate 정보는 PDBx/mmCIF, PDB, XML format 에 저장되어 있다.
(본 글에서는 가장 자주 사용되는 PDBx/mmCIF format과 PDB format 에 대해서만 다루었다)
Atomic level data
•
PDBx/mmCIF format
일종의 key-value 형식으로 여러 정보가 기록되어 있다.
참고) Biopython의 MMCIF2Dict 로 읽으면 실제로 dictionary 형태로 접근이 가능하다.
예시
•
PDB format
각 line의 특정 위치에 어떤 정보를 기록하기로 이미 사전에 정의가 되어 있어, 그 규칙대로 기록되어 있다.
예시
Chain & Models
•
Biological molecule의 hierarchy:
atom → residue → chain → assembly (오름차순 나열)
•
PDBx/mmCIF file format
Chain: 서로 다른 chain은 _atom_site.label_asym_id 라는 category (record)에 표시된다.
예시
Model: 서로 다른 model은 _atom_site.pdbx_PDB_model_num 이라는 record에 표시된다.
예시
•
PDB file format
Chain: 서로 다른 chain은 ‘TER’ 이라는 record를 통해서 한 번 더 명확히 구분해준다.
예시
Model: MODEL/ENDMDL keyword를 통해 구분한다.
예시
Temperature Factors (or B-factor or disorder factor)
•
Atom의 mobility 또는 thermal vibration을 나타내는 수치로, atom 위치의 uncertainty나 flexibility를 어느 정도 알 수 있다.
•
high B-factor (>50): unstable
•
low B-factor (<10): relatively stable
•
PDBx/mmCIF file format
_atom_site.B_iso_or_equiv 에 기록되어 있다.
예시
•
PDB file format
각 line의 61-66 column 에 기록되어 있다.
예시
Occupancy and Multiple Conformations
•
Macromolecular crystal 구조는 일반적으로 실험적으로 얻게 되고, 이 때 대칭적인 구조로 형성이 되므로, 한 사진에서 여러 개가 찍힐 수 있다. 특히 sidechain이나 substrate 같은 경우 다양한 위치에 존재할 수 있다. 이런 형태를 정량적으로 평가한 점수를 occupancy (0~1)라고 한다.
e.g. 어떤 atom이 계속 한 곳에 존재했다면(또는 그렇게 생각해도 무방하다면) 1로 기록
e.g. 어떤 metal ion이 단백질의 특정 부위에 붙은 경우를 세어 봤을 때 해당 위치에 존재하는 빈도가 절반이라면, occupancy는 0.5이다.
•
Occupancy는 side chain이나 ligand가 여러 conformation으로 존재할 경우에도 나타낼 수 있는데, 이 경우 molecule의 fraction을 나타내므로 더해서 1이 되도록 occupancy를 적는다.

편의상 한 conformation만 선택하는 경우가 있는데, 이 방법이 좋지 않을 수도 있으니 주의!
•
PDBx/mmCIF file format
_atom_site.occupancy 위치에 해당 좌표를 가지는 atom의 occupancy가 기록되어 있고, 그 값이 1이 아닐 경우에는 _atom_site.label_alt_id 에 의해 identifier가 기록되어 있어 구분할 수 있다.
예시
•
PDB file format
각 line의 55-60 column 위치에 해당 좌표를 가지는 atom의 occupancy가 기록되어 있고, 그 값이 1이 아닐 경우에는 17 column 위치에 identifier가 기록되어 있어 구분할 수 있다.
예시
Biological Assemblies and the PDB Archive
PDB 홈페이지에서 단백질을 보면, biological assembly 또는 asymmetric unit 이라는 그림을 볼 수 있다. 이 둘의 차이가 무엇일까?
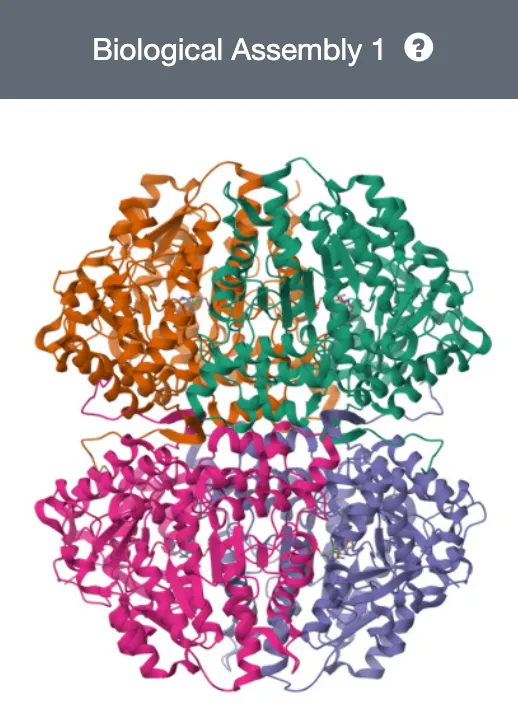
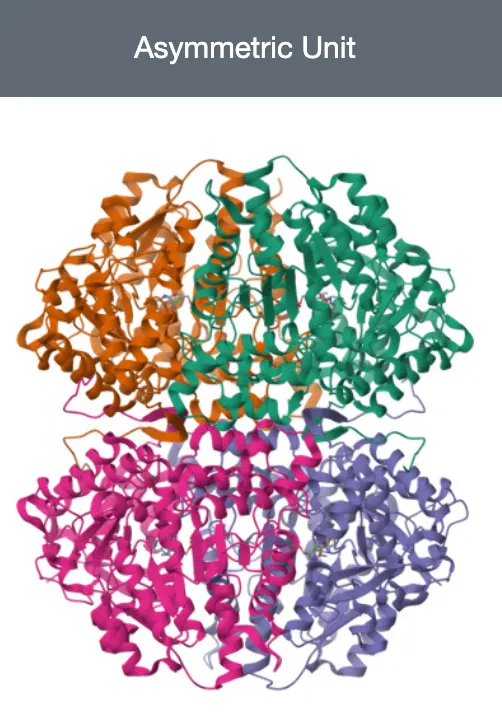
Asymmetric Unit
•
Asymmetric Unit은 symmetry operation이 적용될 수 있는 unit cell을 만드는 가장 작은 portion 이다. 즉, crystal 구조를 구성하는 가장 작은, 반복되는 unit 이다.
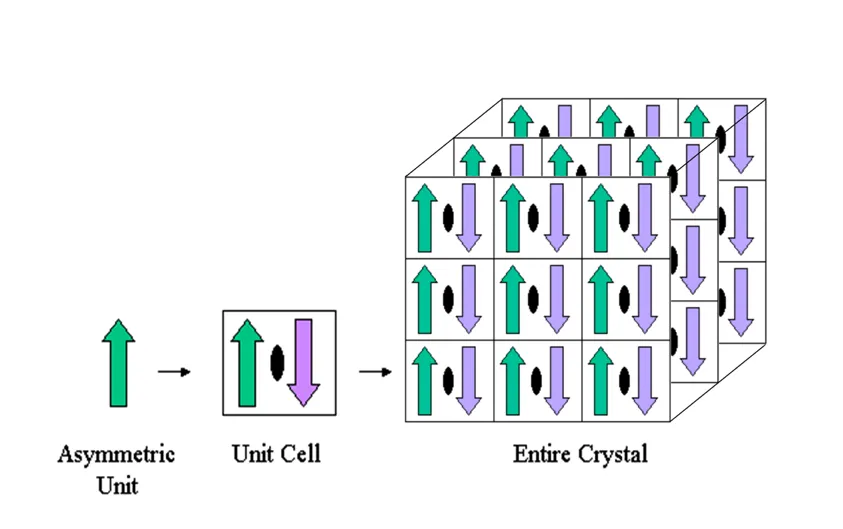
•
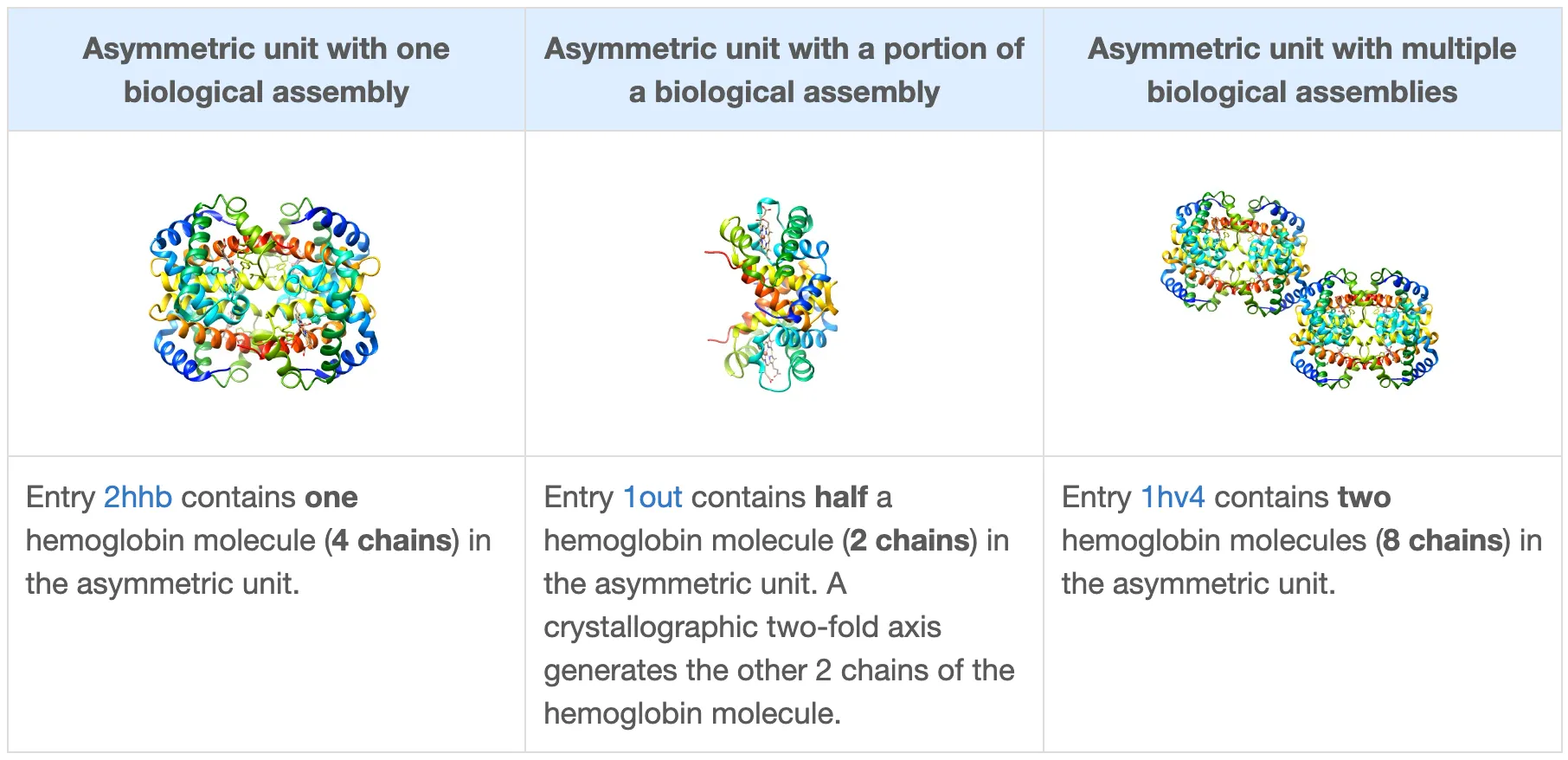
하나의 asymmetric unit은 아래를 포함할 수 있다.
◦
biological assembly 1개
◦
한 biological assembly의 일부분
◦
여러 biological assemblies
Biological Assembly
•
Macromolecular complex의 biologically relevant functional form을 의미한다. 즉, 단백질의 4차 구조이다.
•
Protein은 다른 protein이나 ligand와 결합하여 functional complex를 이룰 수 있는데 이런 것을 biological assembly 형태로 표현한다.
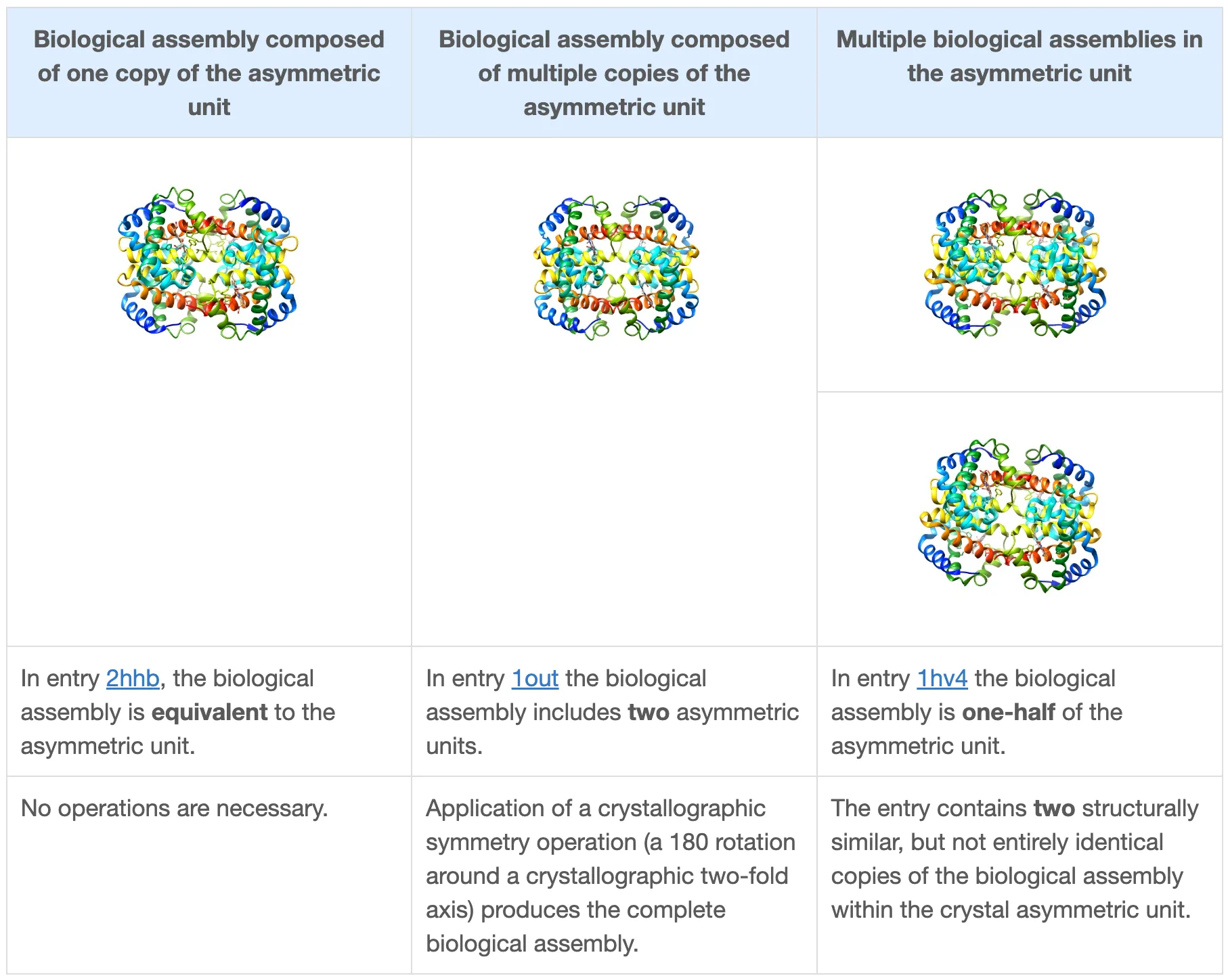
Asymmetric unit vs Biological assembly
•
Asymmetric unit: crystal 구조에서 어떻게 packing 되는지 알 수 있는 구조
•
Biological assembly: functionally meaningful complex의 구조
•
둘이 같을 수도, 다를 수도 있다.
Missing Coordinates and Biological Assemblies
•
어떤 경우엔 실험적인 방법으로 atom을 관측할 수 없는 경우가 있다.
참고)
◦
일반적으로 H atom 은 X-ray crystallography로는 관측되지 않아서, 이 방법으로 얻어진 구조에는 보통 H atom 정보가 없거나 있더라도 별도의 컴퓨터를 이용한 post-processing 된 정보가 기록된다.
◦
Flexible한 region은 실험적으로 관측 자체가 어려운 편이다.
•
모종의 이유로, molecule의 일부분의 좌표만 파일에 포함된 경우도 있다.
참고)
◦
Symmetrical molecule의 X-ray crystallography 구조에 대해, PDB에는 subunit만 있는 경우가 있다. 이 경우, biological assembly의 full coordinate은 subunit을 적절히 rotation (reflection 포함), translation 해서 얻어낼 수 있다. 이렇게 별도의 조립을 해야 하는 경우에는 rotation matrix와 translation vector는 파일에 기록되어 있다.
Coordinate Files
•
실험을 통해서는 낮은 resolution의 단백질 이미지만 얻을 수 있는 경우가 있다. 이 경우, 모든 backbone 또는 side chain atom 들의 좌표를 알 수는 없더라도 일부 (보통은 ) atom 정보만 있는 경우가 있다.
Missing Loops and Tails
•
Flexible region의 경우 X-ray 구조에서 잘 관측이 안 된다. 이런 경우에 chain에 break가 생긴다.
•
NMR로 얻은 구조에서는 이런 문제가 잘 발생하지 않는다. NMR은 여러 구조를 모두 제공 받을 수 있어서, 그 중에 선택해서 사용하거나 전부 사용하면 된다.
이렇게 flexible한 부분이 사실 active site나 binding site에 있는 경우가 종종 있어서, 단순히 무시하고 사용하면 안 된다. 만약 이 부분이 정말 중요한 부분일 경우 비교적 stable conformation을 유지하는 ligand binding state 등을 찾아보면 도움이 될 수 있다.
Fragments and Domains
•
크기가 큰 protein의 경우, 전체를 crystallize하는 것이 어려울 수 있다. 따라서 부분마다 따로 crystallize 하여 database에 올려놓는데, 이들을 잘 조합해서 사용할 수 있다.
PDB entry를 검색할 때, ‘ligand binding domain’ 또는 ‘fragment’ 등의 단어가 제목에 있다면 어떤 단백질의 일부분이라는 것을 유추할 수 있다.
Where are the Hydrogen Atoms?
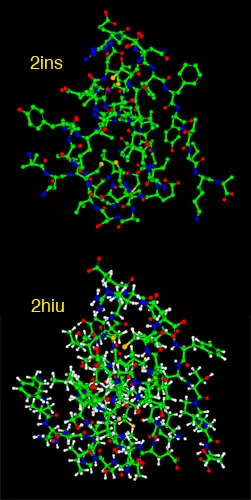
•
대부분의 crystal 기반 구조는 H atom을 찍을 수 없어서, 대부분의 구조에 non-hydrogen atom의 좌표만 있다. N, O, S 등에 결합된 polar hydrogen (수소 결합에 참여하는) 의 경우 좌표가 주어지기도 한다.
•
반면, NMR로 얻은 구조는 H atom의 위치를 알 수 있다.
•
왼쪽 그림은 insulin을 X-ray (위), NMR (아래)로 찍은 그림이다.
•
참고) O와 N의 전자 밀도가 비슷해서 X-ray로는 이 둘을 구분하기 어려운 경우들이 있다. 이로 인해 Asparagine 이나 Glutamine에서 side chain atom을 구별하기 어려운 경우가 있다.
Primary Sequences
•
PDBx/mmCIF file (.cif) 이나 PDB file (.pdb) 에 존재하는 polymer chain (주로 amino acid 또는 nucleotide) 의 경우, 해당 chain의 서열인 primary sequence 정보도 기록되어 있다.
◦
PDBx/mmCIF format
_entity_poly.pdbx_seq_one_letter_code 또는 _entity_poly.pdbx_seq_one_letter_code_can 에 one-letter code 형태로 기록되어 있다.
예시
◦
PDB format
SEQRES (sequence residue) record에 three-letter code 형태로 기록되어 있다.
예시
•
특정 residue의 경우 atom의 좌표가 1개도 찍혀있지 않으면 atom-level 정보에는 해당 residue의 흔적이 없을 수 있다.
이렇게 missing residue가 있더라도 서열 정보는 _entity_poly.pdbx_seq_one_letter_code_can (.cif) 또는 SEQRES record (.pdb) 에 기록되어 있으므로 잘 참고해서 사용해야 한다.
참고) missing residue의 경우는 PDB file의 header 부분의 ‘REMARK 465’ record에 적혀있다.
PDB format에 있는 다른 record들
Record Name | Describes |
MODRES | Modifications to standard residues |
HET | Nonstandard residues (as well as ligands, ions and water) |
HETNAM | Full chemical name of the residue |
HETSYM | Synonyms for the residue |
FORMUL | Chemical formula of the residue |
예시
Hierarchical Structure of Proteins
Myoglobin의 3차 구조가 처음 밝혀지고 나서, 많은 단백질들의 구조가 알려졌다.
단백질의 구조는 hierarchical structure를 가지고 있다고 알려져있다.
참고) Intrinsically Disordered Proteins (IDP)
Why is Understanding Protein Folding Important?
단백질의 기능은 그 구조에 있고, 특정 pocket에 결합할 수 있는 ligand가 존재하는 등, 구조가 매우 중요한 역할을 하기 때문에 1차 구조가 어떻게 3차원적으로 접히는지 알아내는 것이 중요하다고 할 수 있다.
Hierarchical Structure
크게 4단계로 구분된다.
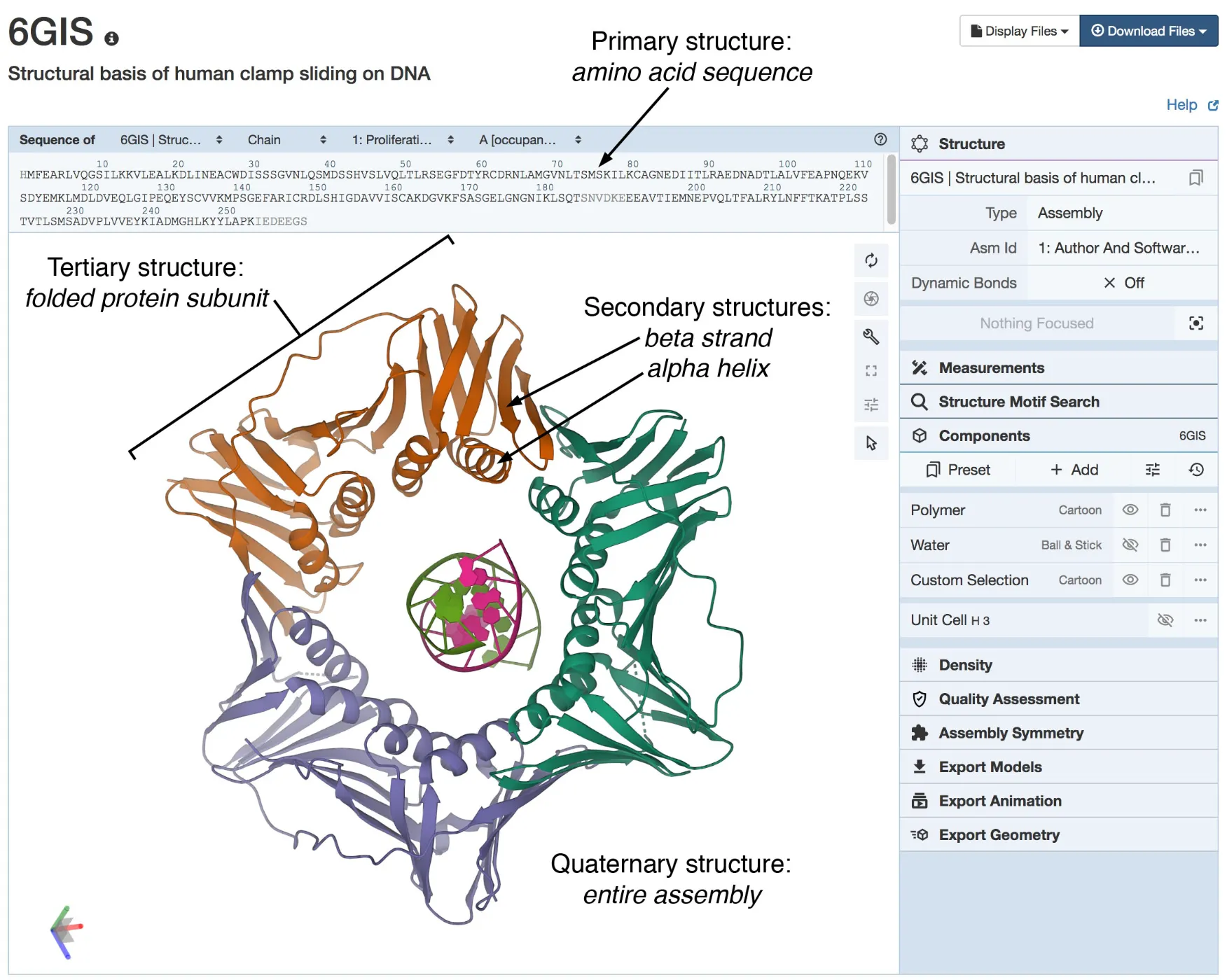
1.
Primary Structure
Protein chain 안의 amino acids 배열 순서. Genome에 encode되어 있고, ribosome에 의해 이 서열이 조합된다.
2.
Secondary Structure
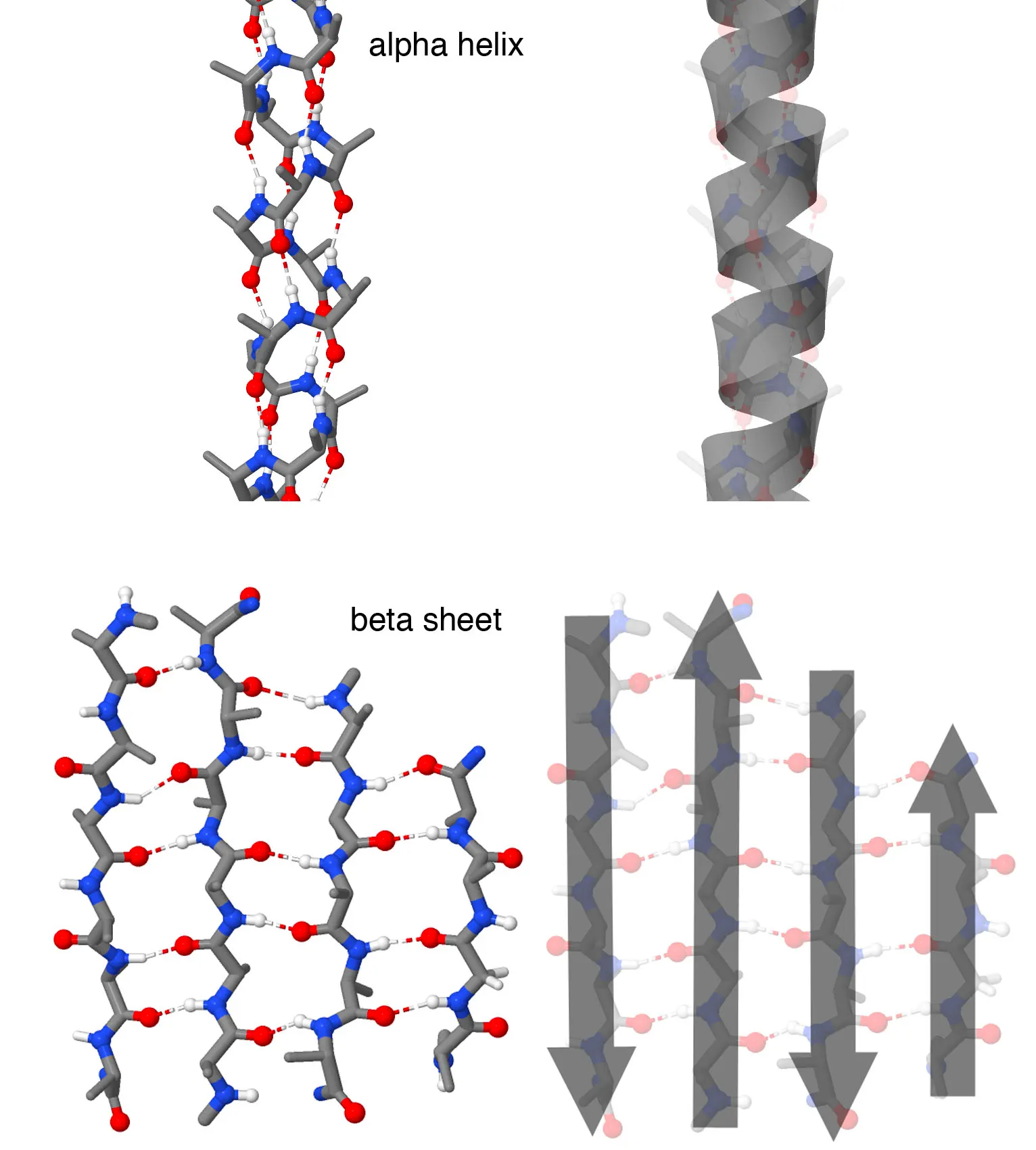
Local region이 특정 형태로 접히는 것을 의미한다. 일반적으로 -helix와 -strand가 존재한다.
이 두 2차 구조 모두 main chain atom이 hydrogen bond를 효율적으로 이루도록 디자인되어 있다.
•
-helix: Helix 내부에서 수소 결합
•
-strand: neighboring strand 간 수소 결합
3.
Tertiary Structure
전체 chain의 2차 구조가 모여 3차 구조를 이룬다.
4.
Quaternary Structure
4차 구조는 두 개 이상의 folded chain이 결합하여 functional assembly를 이룬 형태를 말한다.
각 단백질은 동일하게 구성(homo-oligomers)될 수도 있고, 다르게 구성(hetero-oligomers)될 수도 있다.
이 단백질들은 특정 interface와 orientation을 구성한다.
PDB archive에서 이 4차 구조를 ‘Biological Assembly’라고 부른다.
Visualization of Hierarchical Structure
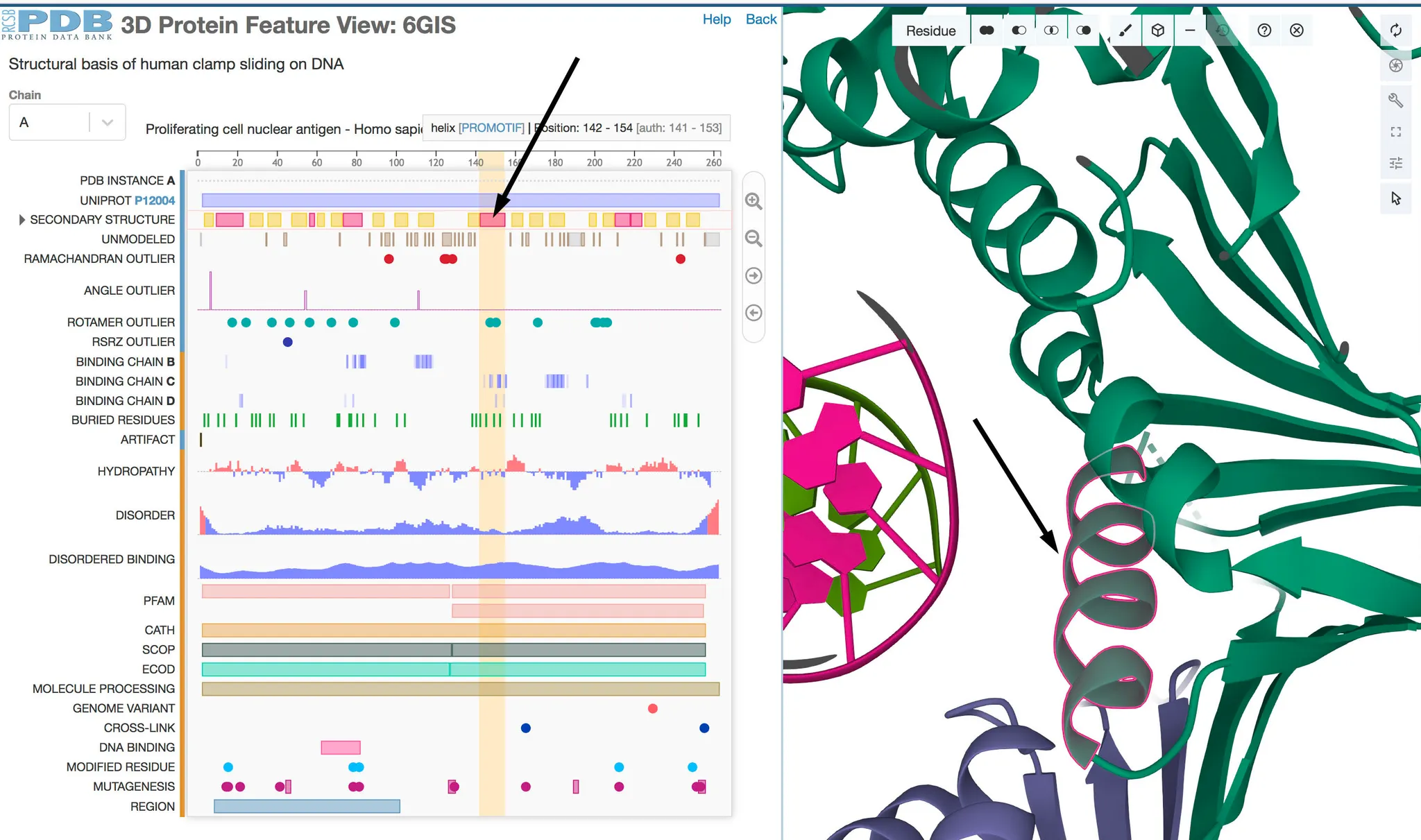
Classifying Protein Folds
여러 단백질 구조가 밝혀지면서, 2차 구조들이 3차 구조로 접히는 특정 규칙이 있다는 것이 발견되었다. 몇 database는 이런 folding을 분류할 수 있도록 했다.
•
CATH
◦
Class(C), Architecture(A), Topology(T), Homologous superfamily(H)를 기준으로 분류.
•
SCOP (Structural Classification of Proteins)
◦
구조적, 진화적 관계를 통해 분류.
Exploring Carbohydrates in the PDB Archive - 나중
형태로 분류되는 sugar, 또는 carbohydrate가 단백질에 결합한 형태로도 존재한다.
이를 또 다른 말로 saccharide라고 부르거나, polymer의 경우 glycan 이라고 부른다.
Glycan은 monosaccharide (glucose 또는 fructose)로 구성된다. 단백질을 구성하는 amino acid는 peptide bond 형태로만 결합하지만, monosaccharide는 다양한 형태로 결합하여 branch를 형성하는 등 여러 구조 형태를 가진다.
Glycan은 protein 또는 lipid에 결합하여 여러 기능을 하므로, 이 구성을 확인하고 이해하는 것이 중요하다.
How Carbohydrates are Specified in PDB Entries
PDB archive에 monosaccharide는 ligand 형태로 간주하고, oligosaccharide는 branded entity로 간주되어 unique chain ID를 가진다.
Small Molecule Ligands
What is a ligand?
많은 biological molecule은 small molecule들 (cofactor, metabolite, or drugs)과 상호작용하고, 이 작은 분자들을 ligand라고 부른다.
참고) wwPDB에서 더 상위 분류로 small molecule과 molecular subunit (amino acid, nucleotide, …)를 모두 포함하는 개념을 “chemical components”라고 명명하였다.
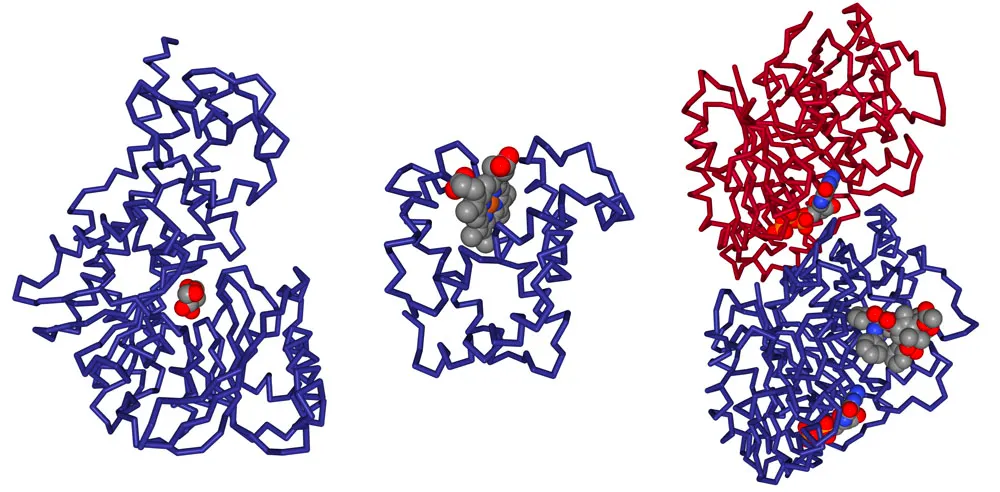
왼쪽) metabolite (glucose)가 hexokinase에 붙음; 중앙) cofactor (heme)이 myoglobin에 붙음; 오른쪽) drug (taxol)이 tubulin에 붙음.
모든 chemical component는 알파벳 3개로 표현한다. 예를 들어, alpha D-glucose는 GLC, heme cofactor는 HEM 으로 표기한다. 자세한 것은 wwPDB Chemical Component Dictionary (CCD)에 소개되어 있다.
따라서 특정 ligand를 찾기 위해서는 PDB에서 이 알파벳으로 검색하면 된다.
Representation of Ligands in Entry Files
Ligand는 mmCIF와 PDB format에서 조금 다른 방식으로 표기되어 있으나, 전달하고자 하는 정보 (characteristic, interaction, coordinate)는 비슷하다.
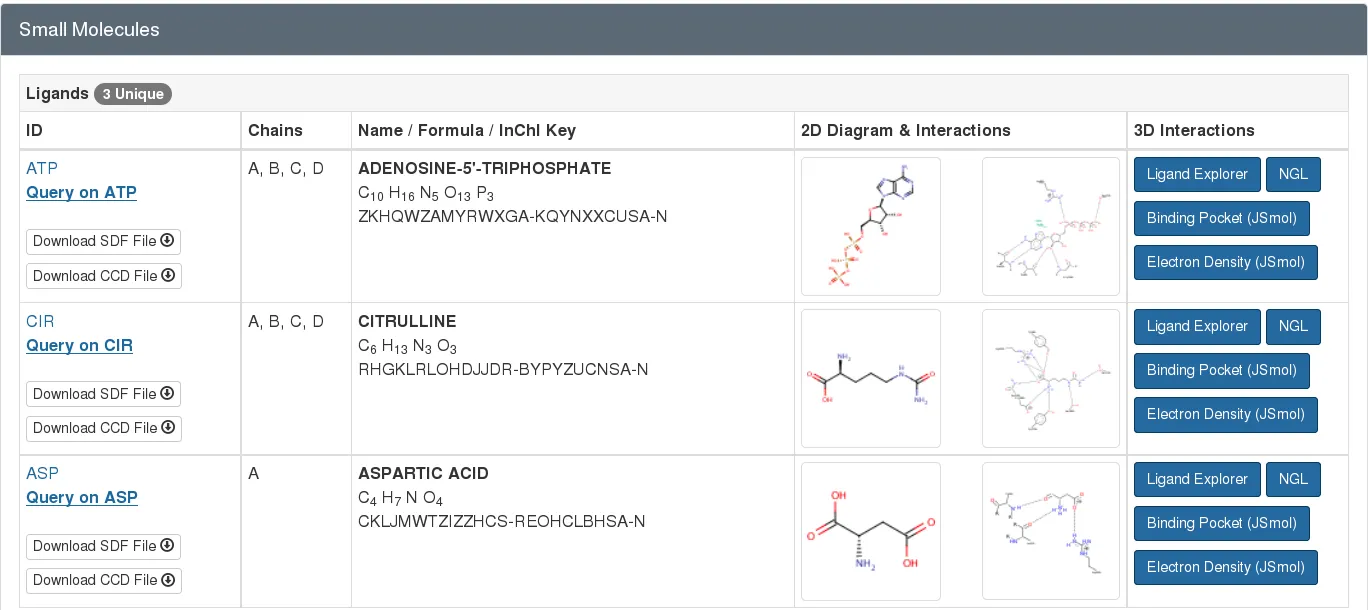
PDB entry 1j1z에 대해 결합할 수 있는 small molecules.
Free Ligands vs. Ligands in Polymers
대부분의 ligand는 macromolecule과 공유결합을 하고 있지 않은 ‘free ligand’ 이지만, 간혹 공유결합을 하는 ‘ligands in polymers’ 인 경우나 아예 macromolecule에 포함되어 있는 경우도 있다. PDB data에서는 이 내용이 구분되어 표기되어 있다.
Amino acid나 nucleotide 같이 protein이나 nucleic acid의 building block인 경우도 PDB archive에서 ligand로 표기되는 경우가 있다. 예를 들어, PDB entry 1j1z에서 aspartate (ASP)는 free ligand로 작동한다.
또한, 주로 unbound state로 발견되는 ligand임에도 polymer chain의 일부분으로 기록되는 경우가 있다. 특히 modified amino acid인 hydroxyproline (HYP) 또는 selenomethione (MSE) 같은 경우가 그렇다.
Free ligand가 공유 결합 또는 금속 결합을 할 경우에도 data에 표현이 된다. 예를 들어, PDB entry 1pwc (penicillin G)는 bacterial enzyme DD-peptidase의 catalytic serine과 결합한다고 알려져 있다.
Biologically Interesting Molecules
wwPDB에서 complex ligand (peptide-like inhibitors, complex antibiotics, ribosomally synthesized gene products, products of nonribosomal enzymatic synthesis) 를 다루기 위해 Biologically Interesting molecule Reference Dictionary (BIRD)를 만들었다. 여기에는 thiostrepton, vancomycin, viomycin 등이 포하모디어 있다.
Molecular Graphics Programs
What does a protein or nucleic acid look like?
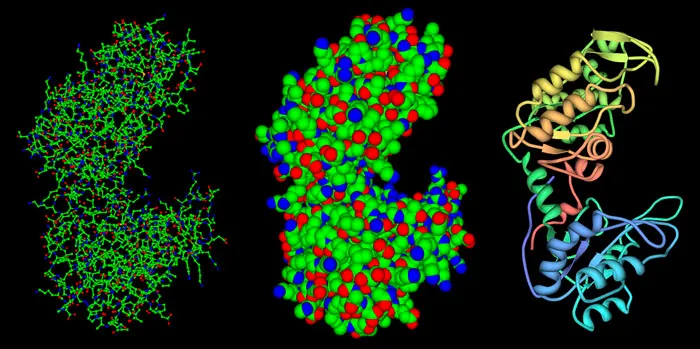
3가지 type
1.
Wireframe Diagrams (왼쪽)
Atom은 small ball, covalent bond는 stick으로 이은 구조.
특정 부분 (active site, metal ion 결합 부위 등)을 구체적으로 보기에 좋다.
2.
Spacefilling Diagrams (가운데)
각 atom에 대해 sphere를 그려서 상대적인 atom size를 비교할 수 있다.
단백질의 전체적인 모양과 서로 다른 단백질들이 어떻게 상호작용 하는지 보기 좋다.
3.
Backbone and Ribbon Diagrams (오른쪽)
Protein chain이 어떻게 접혀있는지 볼 수 있다. 2차 구조 (-helix, -strand 등)을 함께 볼 수도 있다.
단백질 접힘 구조를 보거나 서로 다른 단백질끼리 비슷하게 접힌 부분을 볼 때 좋다.
Selection and Coloring
단백질이 매우 복잡하기 때문에 특정 부분을 선택해서 색칠하면 보기 편하다.
Methods for Determining Structure
단백질 구조를 결정하는 방법은 크게 3가지가 있다: X-ray crystallography, NMR spectroscopy, Electron microscopy. 이 방법들로 찍기만 하면 바로 구조를 알 수 있는 것은 아니고, 이미 알려진 지식들 (amino acid squence, bond length, bond angle 등)을 조합해서 구조를 알아낸다.
X-ray crystallography
PDB archive에 있는 대부분의 구조는 X-ray로 얻었다. 단백질을 정제하고 결정화하여 X-ray beam을 여러 차례 쏜다. 결정 안의 단백질이 X-ray beam을 회절시켜 어떤 점의 pattern을 만들어낸다.
PDB archive에는 crystal 구조 데이터가 두 종류 있다.
•
Atomic position을 담고 있는 coordinate file
•
Structure factor (X-ray의 intensity나 phase 등)를 담고 있는 file.
X-ray가 굉장히 많은 정보를 줄 수 있지만, 단백질을 결정화 하는 것이 매우 어렵고, 결정화가 아예 안 되는 단백질도 있다. Rigid한 구조를 가지는 단백질에 대해서는 굉장히 좋지만, flexible한 단백질은 X-ray로 제대로 찍기 어렵다. 동일한 구조가 반복되어야 일정한 회절 패턴을 관측할 수 있기 때문이다.
Crystallographic structure의 accuracy를 측정하는 두 가지 중요한 metric:
•
resolution: 실험 데이터에서 얼마나 detail까지 볼 수 있는지
•
R-value: structure factor file이 atomic model을 얼마나 잘 설명하는지
NMR spectroscopy
단백질을 정제하여 강한 자기장에 넣고 radio wave를 주면 원자들이 서로 다른 공명 패턴을 보이는데, 이를 분석해서 구조를 알아낸다. 현재까지는 작거나 중간 사이즈 정도의 단백질에만 적용할 수 있다. 큰 단백질의 경우 NMR spectrum이 겹치게 나타나서 분석이 어렵다.
NMR 구조의 큰 장점은 억지로 만든 결정이나 microscrope grid에 잡히지 않은, solution 환경에 존재하는 단백질의 구조를 알 수 있다는 점이다. 따라서 flexible protein의 경우 NMR로 얻은 구조를 가장 신뢰한다.
PDB archive에 NMR 관련 데이터가 두 종류 있다.
•
Full ensemble: 각 구조가 독립적인 model로 표시되어 있음
•
Minimized average: 모든 구조의 average?
Electron microscopy (3DEM)
Biomolecule에 전자 beam을 직접 쬐어 구조를 관측한다. Cryo-EM을 사용하면 얇은 non-crystalline ice 상에 Biomolecule이 여러 각도로 존재하고, 전자 beam을 쏘아 2D 형태로 projection 된 구조를 컴퓨터로 재구성하여 구조를 알아낸다.
최근 3DEM 기술이 발전하면서 crystallography에 맞먹는 resolution을 보여주고 있다. Cryo-electron tomography 라는 기술은 좀 더 낮은 resolution에서 protein의 domain이나 secondary structure를 찍는데 사용하고 있다.
큰 macromolecule의 경우, 3DEM data, X-ray, NMR 등 여러 data를 함께 사용하여 구조 결정에 활용한다.
Integrative Modeling
더 크고 복잡한 생물학적 시스템을 연구하기 위해, 다양한 data source를 활용하는 integrative modeling 이라는 기법이 활용되고 있다.
예를 들어, nuclear pore complex (NPC)의 구조는 integrative modeling으로 알게 되었다. 먼저 electron microscopy로 전체적인 구조를 파악하고, chemical crosslinking 실험을 통해 상대적인 위치와 orientation을 파악했다.
Computed Structure Models
Protein의 1차 구조 (sequence)가 functional 3D shape을 결정한다. 따라서 이 둘의 관계를 잘 모델링한다면 functional shape을 알 수 있을 것이다. 그러나 정확히 어떤 양상으로 접히는지 그동안 제대로 파악하지 못 했다.
안정적으로 접힌 단백질은 free energy가 가장 낮은 상태라고 생각된다. 따라서 protein structure prediction 도 polypeptide chain이 안정적으로 상호작용 하여 free energy를 최소화도록 해야 한다.
하지만 이런 계산은 매우 계산양이 많아, physics-based software tool (Rosetta 같은)은 짧은 polypeptide chain에 대해서만 수행한다. 큰 단백질에 대해 구조를 계산할 경우, 추가적인 정보가 필요하고 그동안 크게 template-based와 template-free modeling 두 분류의 방법들이 제안되어 사용되었다.
Template-based modeling
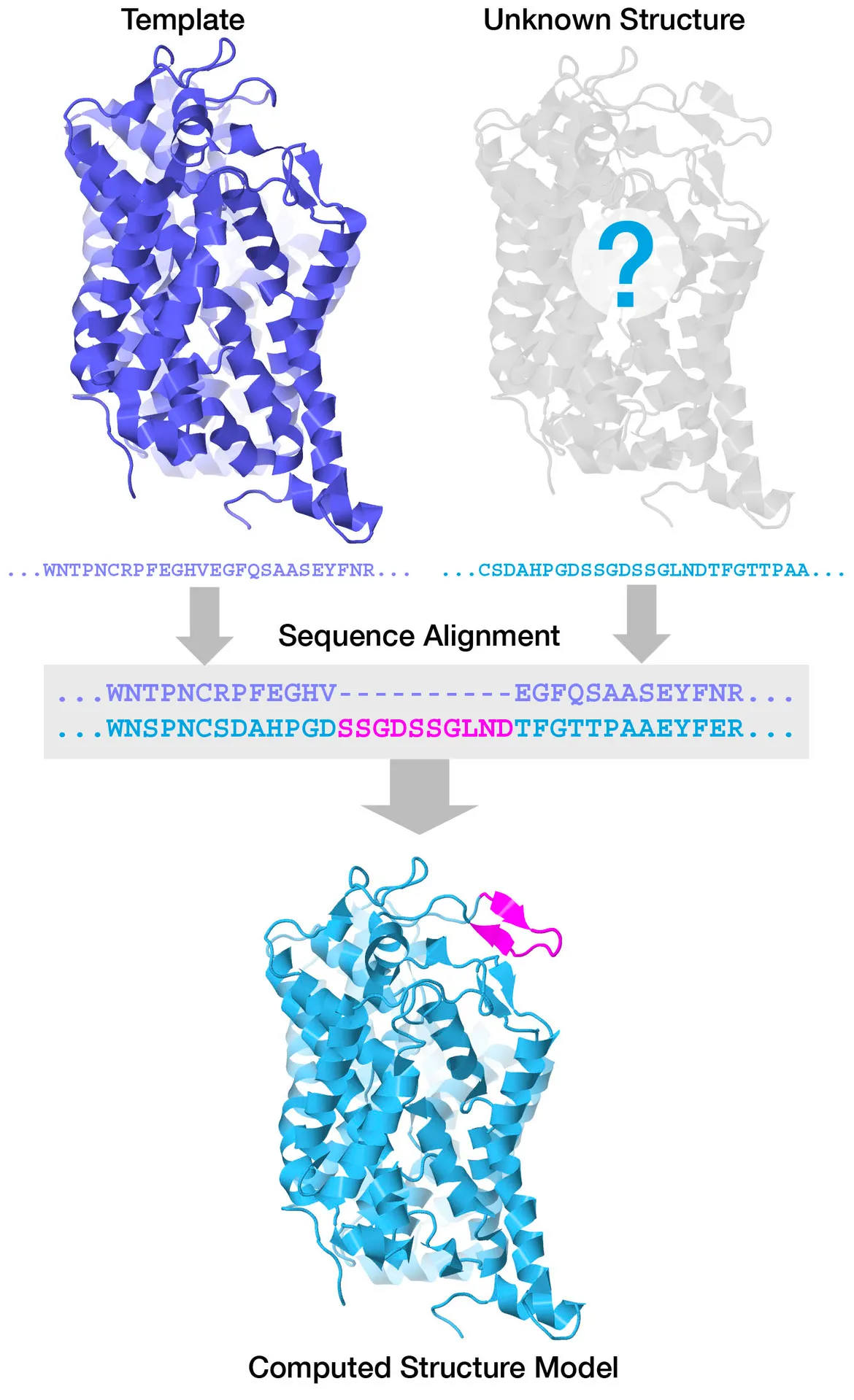
서로 비슷한 아미노산 서열(sequence identity 30% 이상)을 가진 단백질은 비슷한 구조로 접힌다는 것이 알려져 있다. 따라서 새로운 단백질의 구조를 알아낼 때, homologous protein의 구조를 활용할 수 있다.
이렇게 비슷한 구조에서 기반한 homology modeling은 sequence identity가 40% 이상일 경우 꽤 잘 작동한다고 알려져 있고, 이미 잘 알려진 구조를 template으로 활용하여 예측에 사용한다.
Template-free modeling
Homologous structure (template)이 없을 경우, local feature를 match하여 모델을 만든다. Multiple sequence alignment를 통해 진화적으로 보존된 부분을 알 수 있다. 3차원 구조 상에서 가까이 위치하는 아미노산 끼리는 그 둘이 mutation이 있더라도 비슷한 interaction을 가지도록 진화했다. 이런 covariance는 distance 상의 constraint를 제공하여 intramolecular contact에 대한 힌트를 제공할 수 있다. 또한, fragment match나 local secondary structure prediction이 template free structure prediction에 활용될 수 있다.
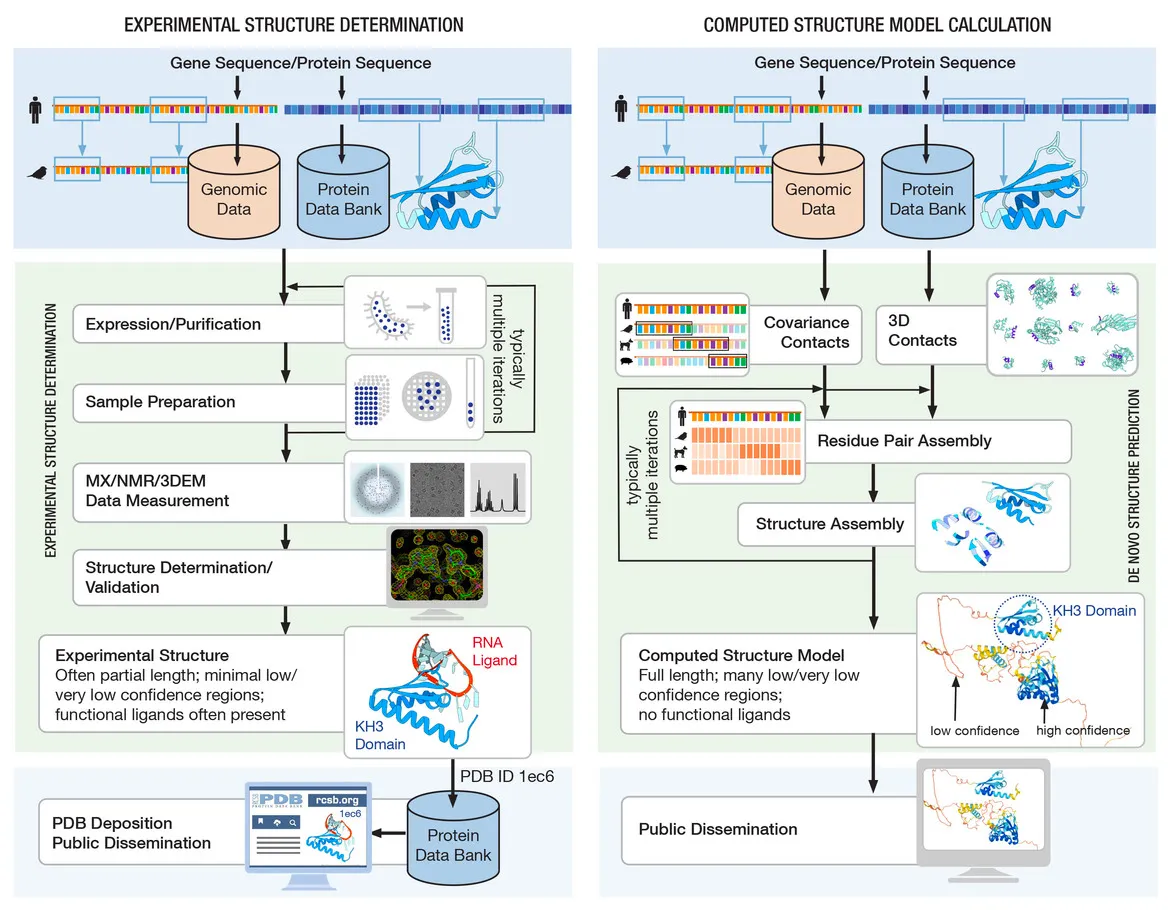
Generating Computed Structure Models
1990년대 초반부터 CASP team이 2년에 한 번씩 computational algorithm을 활용한 구조 예측 대회를 개최하였다. 2020년에 AlphaFold2 (AF2)가 AI와 ML을 활용해 높은 정확도로 예측하였고, 뒤이에 RoseTTAFold 도 개발되었다. 이 두 방법 모두 기존에 알려진 sequence data와 3D 구조를 활용하였다.
AF2는 다음과 같은 순서로 구조를 예측한다.
1.
구조를 알고 싶은 단백질의 서열에 대해 multiple sequence alignment를 수행한다.
2.
Iterative process로 MSA를 활용하여 구조를 refine 해나간다.
최종 모델은 predicted Local Distance Difference Test (pLDDT)라고 하는 confidence score도 예측하여 어떤 예측에 대해 얼마나 확신하는지도 제공한다.
Using Computed Structure Models (CSMs)
아직까지 AF2나 RF 같은 단백질의 3D 구조 예측 모델은 잘 접히고, rigid하고, higher-order 구조를 형성하지 않는 단백질에 대해서 매우 잘 수행한다. 이 모델들은 PDB에 없는 단백질의 중요성을 테스트해보거나, 구조 기반의 약물 개발에 사용하거나, 3D 구조 결정 자체에 활용될 수 있다.
그렇지만 어떤 단백질의 구조가 PDB에 이미 잘 정의되어 있다면, 그것을 바로 활용하는 것이 좋다 (CSM을 돌리기 보다는).
AlphaFoldDB, ModelArchive 등에 수백만개의 단백질에 대해 구조를 예측한 결과가 업로드 되어 있으니, 이를 활용해보는 것도 좋다.
Case Study: Computed Structure Model of the Src Oncoprotein
실제로 단백질은 매우 dynamic한 구조이다. 주변 환경에 따라 매우 다른 conformation을 가질 수도 있어서 하나의 구조만으로 그 기능을 모두 설명할 수 없다.
Src oncoprotein은 현재 AI/ML tool의 장점과 한계를 모두 보여주는 예시이다. Human Src는 multi-domain protein으로, flexible한 N-terminal tail, SH3 domain, SH2 domain, kinase domain, small C-terminal tail로 구성되어 있다. Src protein이 active 할 때는 단백질의 세 domain이 모두 서로 떨어져있고, kinase domain이 cell 안에 위치하여 target protein에 대해 phosphate group을 추가하는 반응을 촉진하도록 한다.
C-terminal의 tyrosine residue가 매우 중요한 역할을 하는데, 다른 protein kinase에 의해 인산화되면 Src protein을 inactive하게 만들 수 있다. SH2와 SH3 domain이 서로 붙어 전체 polypeptide chain을 compact하고 닫힌 구조로 만든다. 실험적으로도 이때의 상태가 compact 하다는 것이 확인된다.
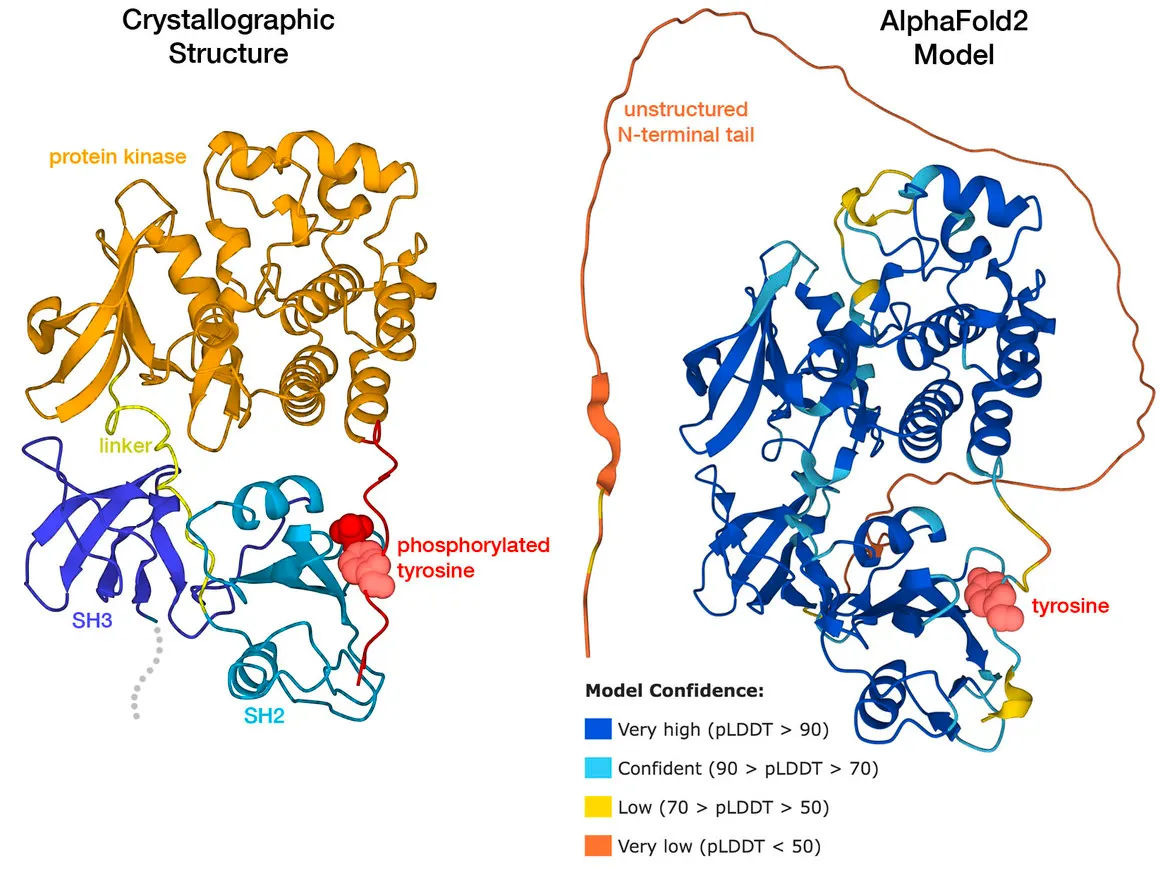
AF2와 RF 모두 세 개의 globular domain에 대해서는 잘 예측을 하지만, active 하고 인산화되지 않은 Src 형태는 잘 못 예측하는 것을 확인할 수 있다.
따라서 이런 CSM의 예측값을 모두 믿기 보다는 적절히 잘 활용하자!
Resolution
TBU
R-value and R-free
TBU
Structure Factors and Electron Density
TBU