

.png&blockId=8315391e-7f90-470c-9153-95e5b093ac8c&width=3600)

•
NeurIPS 2021 Poster
Summary
•
Used physicist network (PhysNet) and chemist network (ChemNet) simultaneously, and each network shares information to solve individual tasks.
•
PhysNet: Neural physical engine. Mimics molecular dynamics to predict conformation.
•
ChemNet: Message passing network for chemical & biomedical property prediction.
•
Molecule without 3D conformation can be inferred during test time.
Preliminaries
•
Molecular representation learning
Embedding molecules into latent space for downstream tasks.
•
Neural Physical Engines
Neural networks are capable of learning annotated potentials and forces in particle systems.
HamNet proposed a neural physical engine that operated on a generalized space, where positions and momentums of atoms were defined as high-dimensional vectors.
•
Multi-task learning
Sharing representations for different but related tasks.
•
Model fusion
Merging different models on identical tasks to improve performance.
Notation
•
Graph
: set of atoms
: set of chemical bonds
: matrix of atomic features
: matrix of bond features
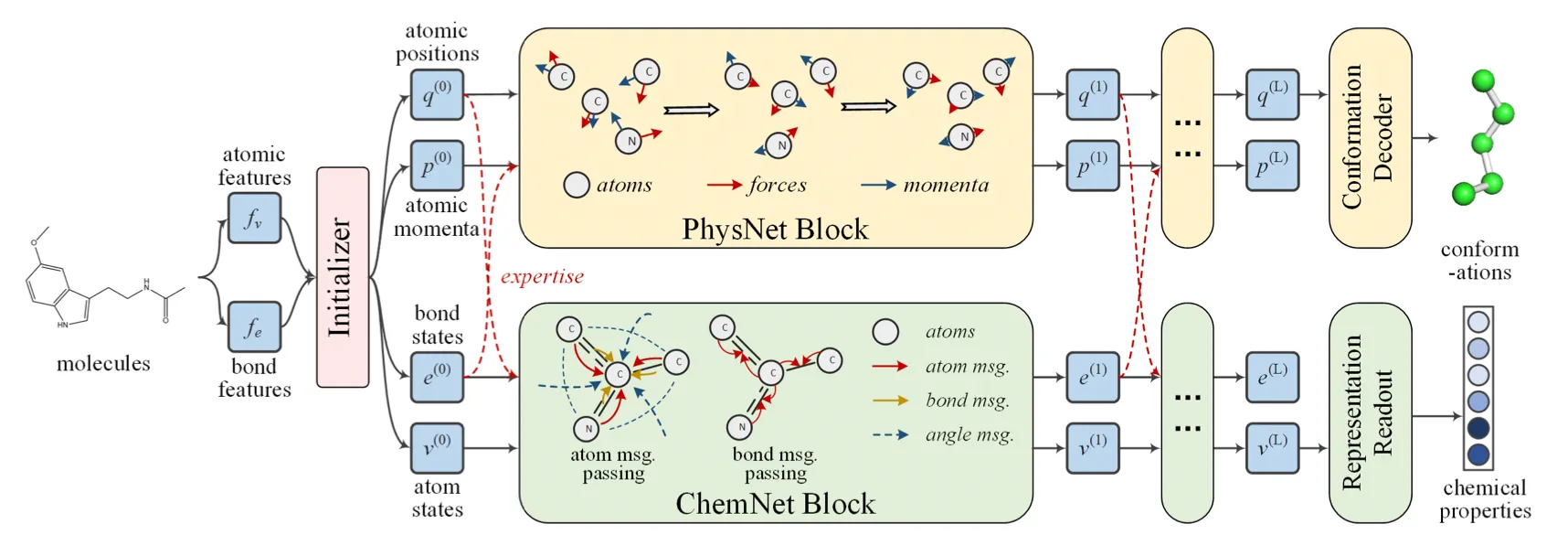
Model
•
Initializer
◦
Input: atomic features, bond features (from RDKit)
◦
Layer: fully connected layers
◦
Output:
▪
bond states, atom states for ChemNet
,
▪
atom positions, atomic momenta for PhysNet
Bond strength adjacency matrix
•
PhysNet
◦
HamNet showed that neural networks can simulate molecular dynamics for conformation prediction.
◦
Directly parameterize the forces between each pair of atoms.
◦
Consider the effects of chemical interactions(e.g. bond types) by cooperating with ChemNet’s bond states.
◦
Introduces torsion forces.
◦
Output: 3D conformation
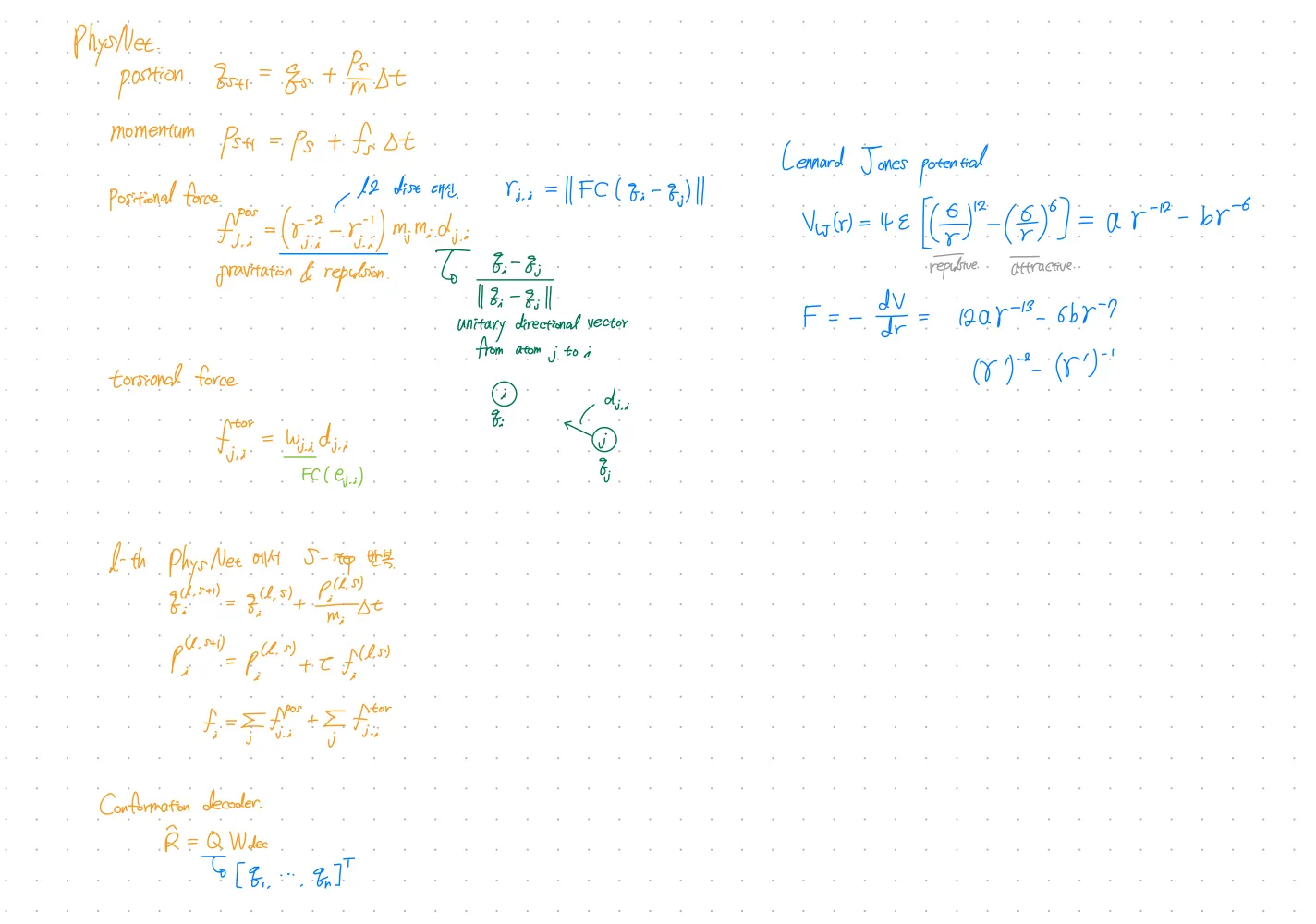
•
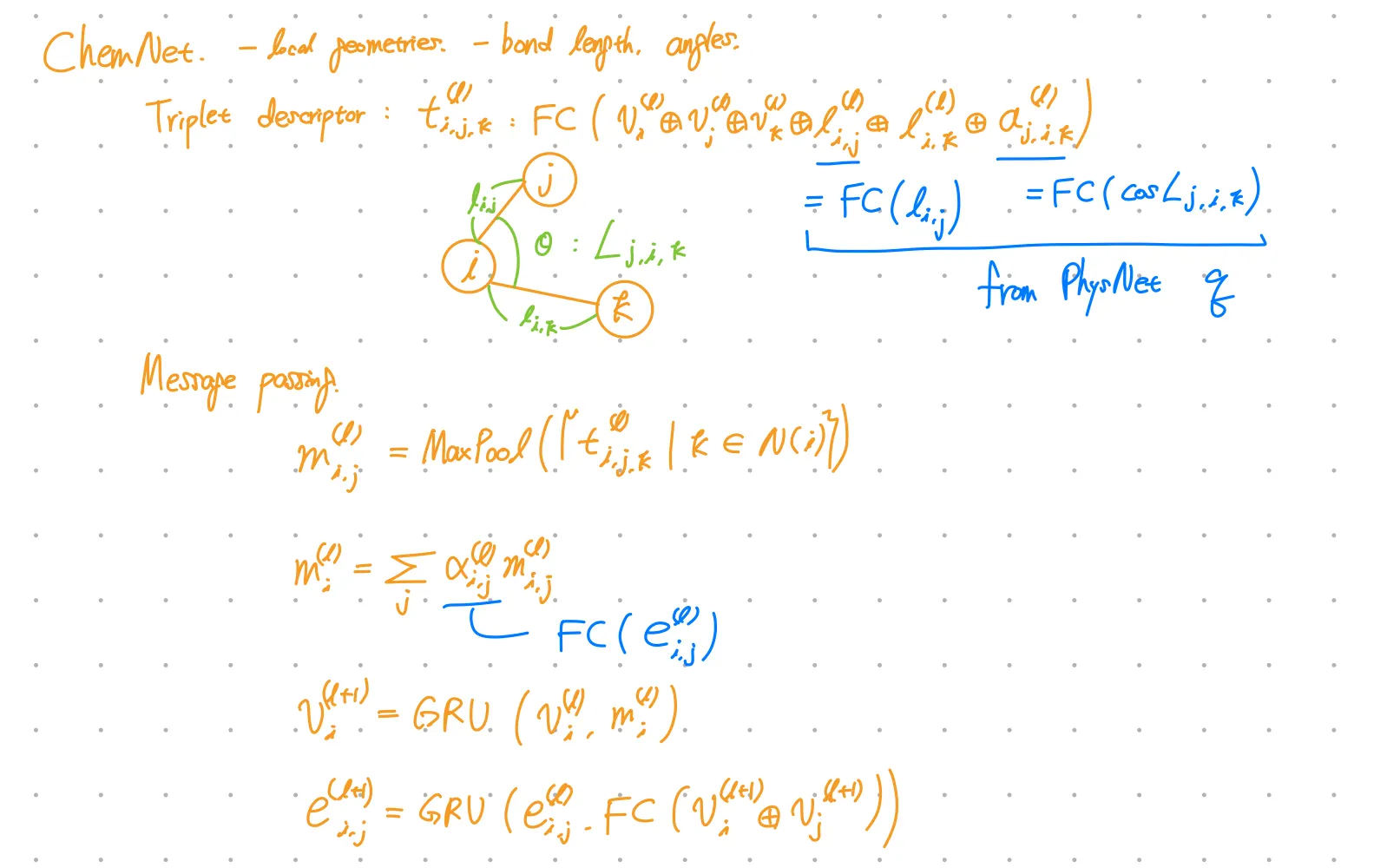
ChemNet
ChemNet modifies MPNN(message passing neural network) for molecular representation learning.
◦
Output: Molecule representation
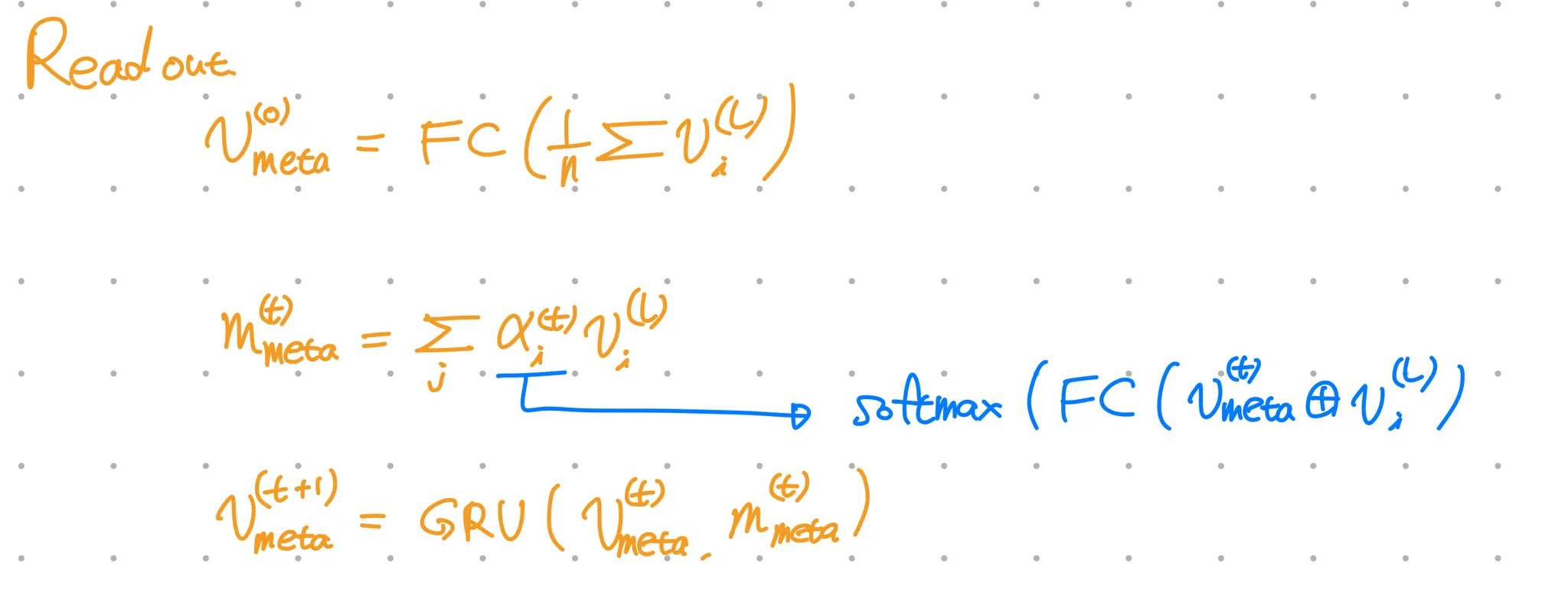
Loss
•
: Conn-k loss for Conformation prediction (PhysNet)
-hop connectivity loss
: element-wise product
: Frobenius norm
: distance matrix of the real and predicted conformations
: normalized -hop connectivity matrix
•
: MAE or Cross entropy loss for Property prediction (ChemNet)
•
Total loss
Checkpoints
•
Is Conn-k loss generally used in other conformation prediction models?
No! But seems related to local distance loss.
•
Is triplet descriptor generally used in other models?
No!