

Table of Contents
•
This article is one of the first research work that formulated molecular docking as a generative problem.
•
Showed very interesting results with decent performance gain.
•
If you are interested in molecular docking and diffusion models, this is definitely a must-read paper!
•
Summary
•
Molecular docking as a generative problem, not regression!
◦
Problem of learning a distribution over ligand poses conditioned on the target protein structure
•
Used “Diffusion process” for generation
•
Two separate model
◦
Score model:
Predicts score based on ligand pose , protein structure , and timestep
◦
Confidence model:
Predicts whether the ligand pose has RMSD below 2Å compared to ground truth ligand pose
•
Diffusion on Product space
◦
Reduced degrees of freedom
Preliminaries
Molecular Docking
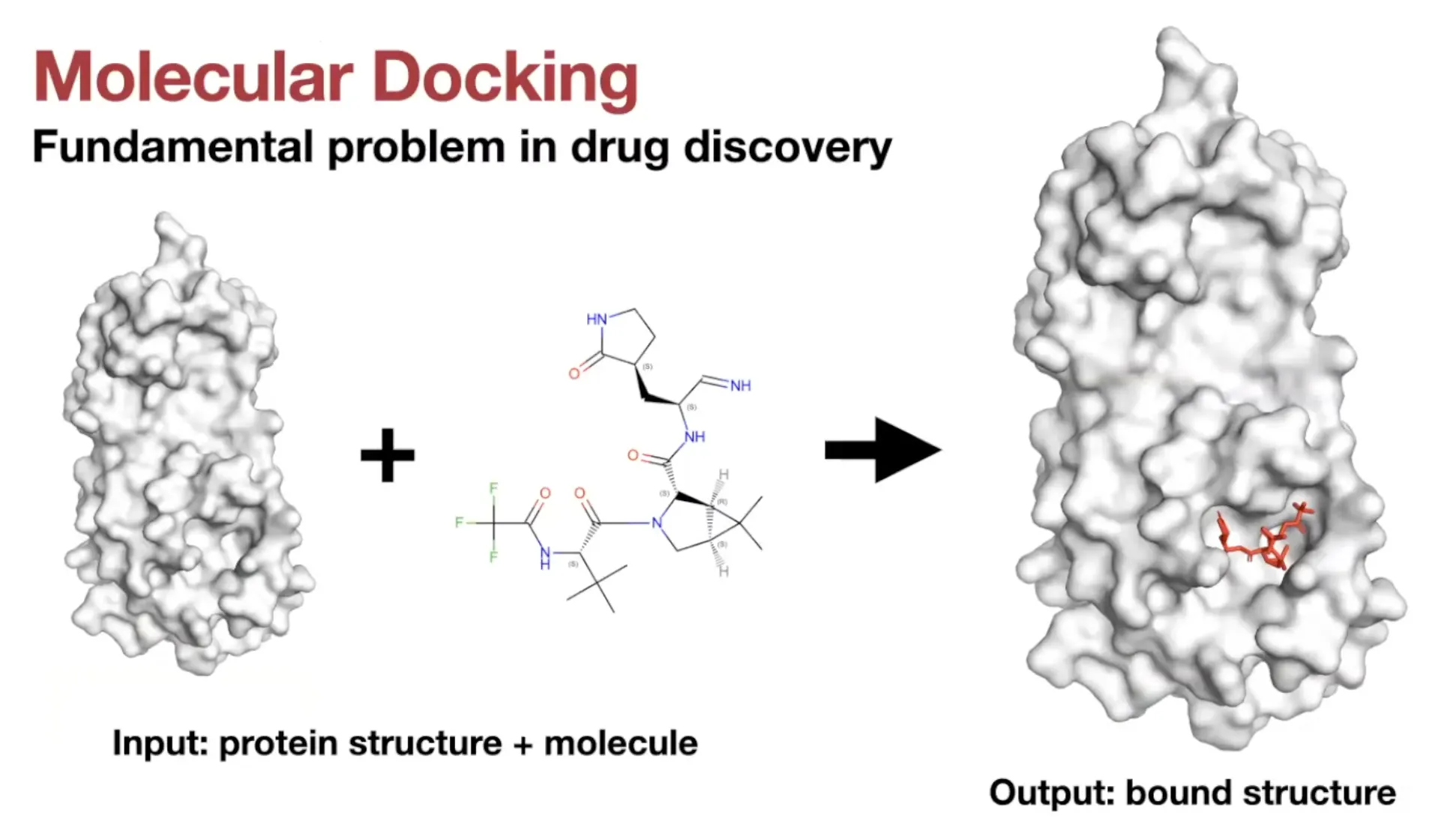
•
Definition:
Predicting the position, orientation, and conformation of a ligand when bound to a target protein
•
Two types of tasks
◦
Known-pocket docking
▪
Given: position of the binding pocket
◦
Blind docking
▪
More general setting: no prior knowledge about binding pocket
Previous works: Search-based / Regression-based
•
Search based docking methods
◦
Traditional methods
◦
Consist of parameterized physics-based scoring function and a search algorithm
◦
Scoring function
▪
Input: 3D structures
▪
Output: estimate of the quality/likelihood of the given pose
◦
Search algorithm
▪
Stochastically modifies the ligand pose (position, orientation, torsion angles)
▪
Goal: finding the global optimum of the scoring function.
◦
ML has been applied to parameterize the scoring function.
▪
But very computationally expensive (large search space)
◦
Example

•
Regression based methods
◦
Recent deep learning method
◦
Significant speedup compared to search based methods
◦
No improvements in accuracy
◦
Example

▪
•
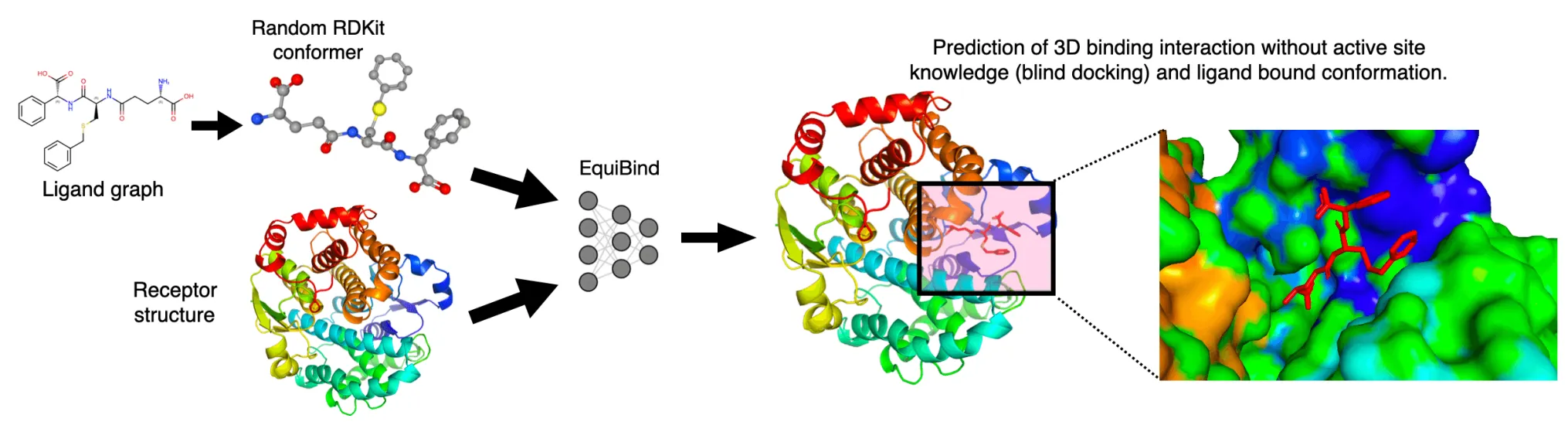
Tried to tackle the blind docking task as a regression problem by directly predicting pocket keypoints on both ligand and protein and aligning them.
▪
•
Improved over this by independently predicting a docking pose for each possible pocket and then ranking them.
▪
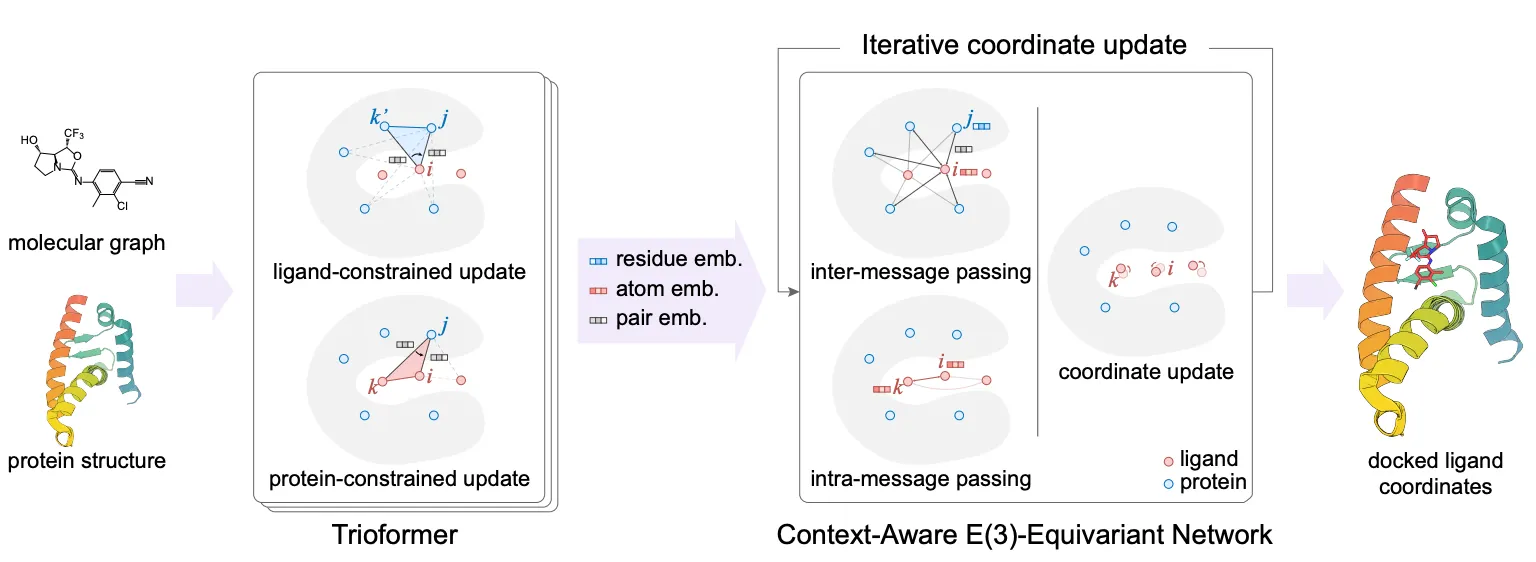
•
Used ligand-constrained & protein-constrained update layer to embed ligand atoms and iteratively updated coordinates.
Docking objective
•
Standard evaluation metric:
◦
: proportion of predictions with → Not differentiable!
◦
Instead, we use as objective function.
•
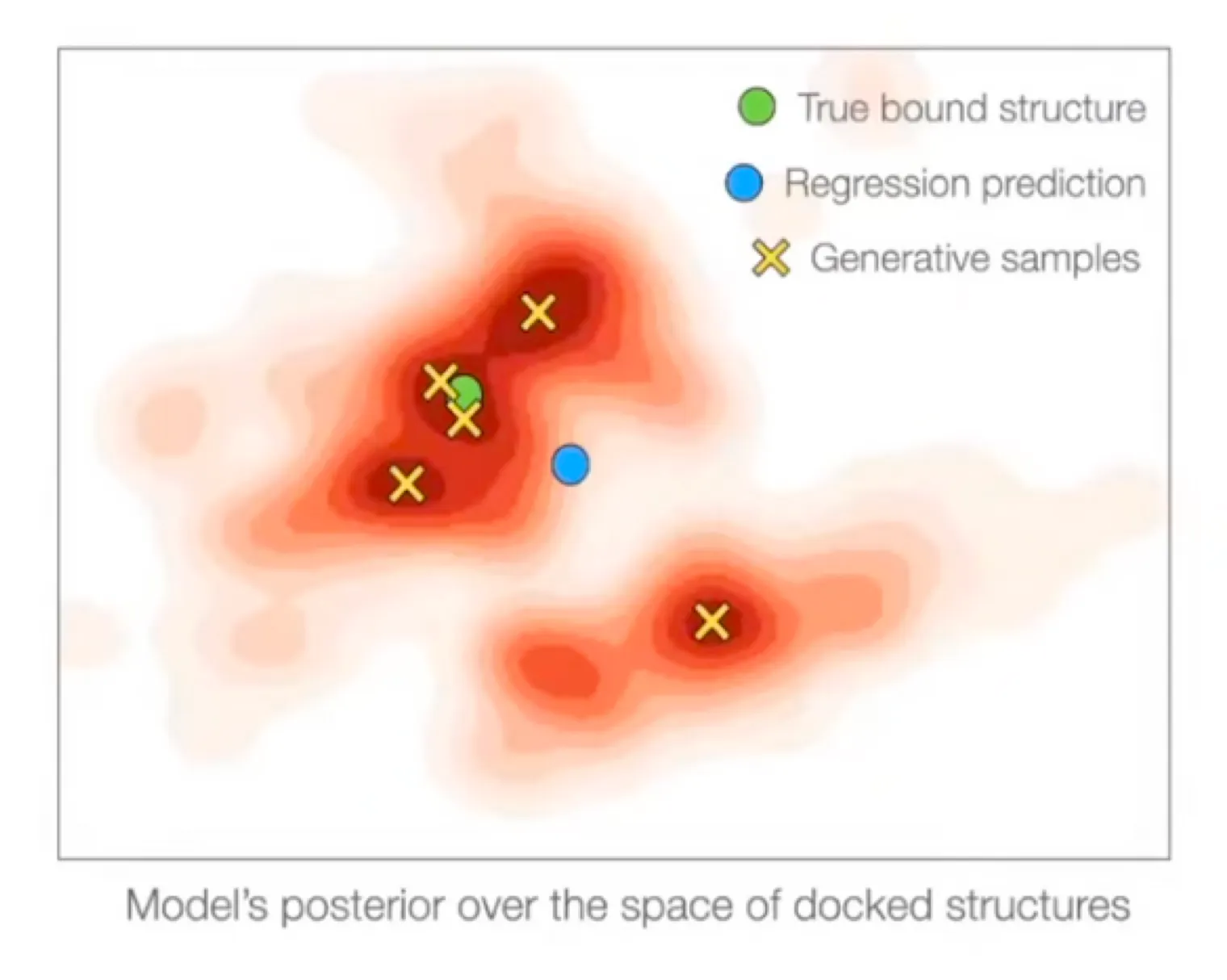
Regression is suitable for docking only if it is unimodal.
•
Docking has significant aleatoric (irreducible) & epistemic (reducible) uncertainty
◦
Regression methods will minimize → will produce weighted mean of multiple modes
◦
On the other hand, generative model will populate all/most modes!
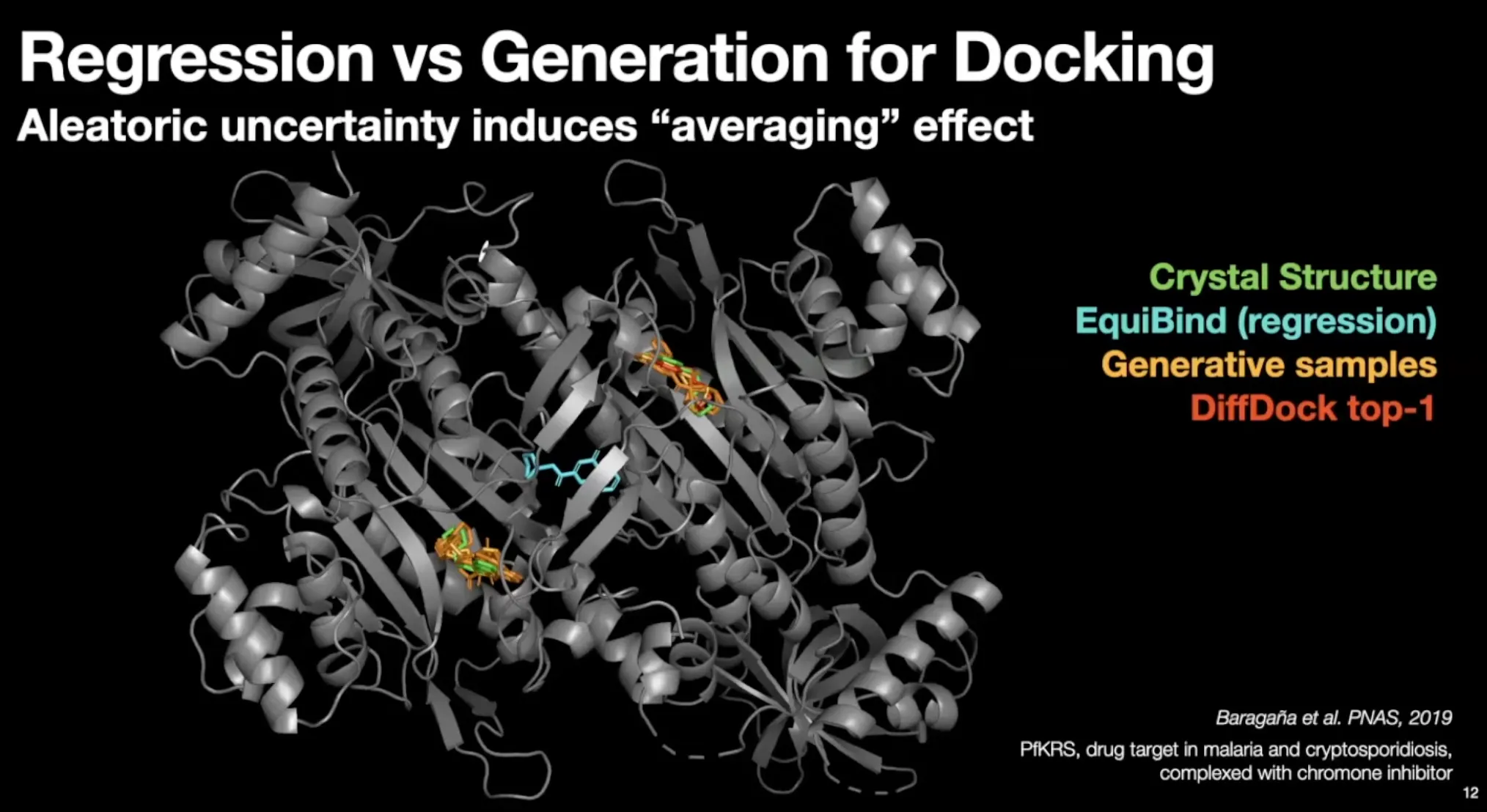
•
Regression (EquiBind) model set conformer in the middle of the modes.
•
Generative samples can populate conformer in most modes.
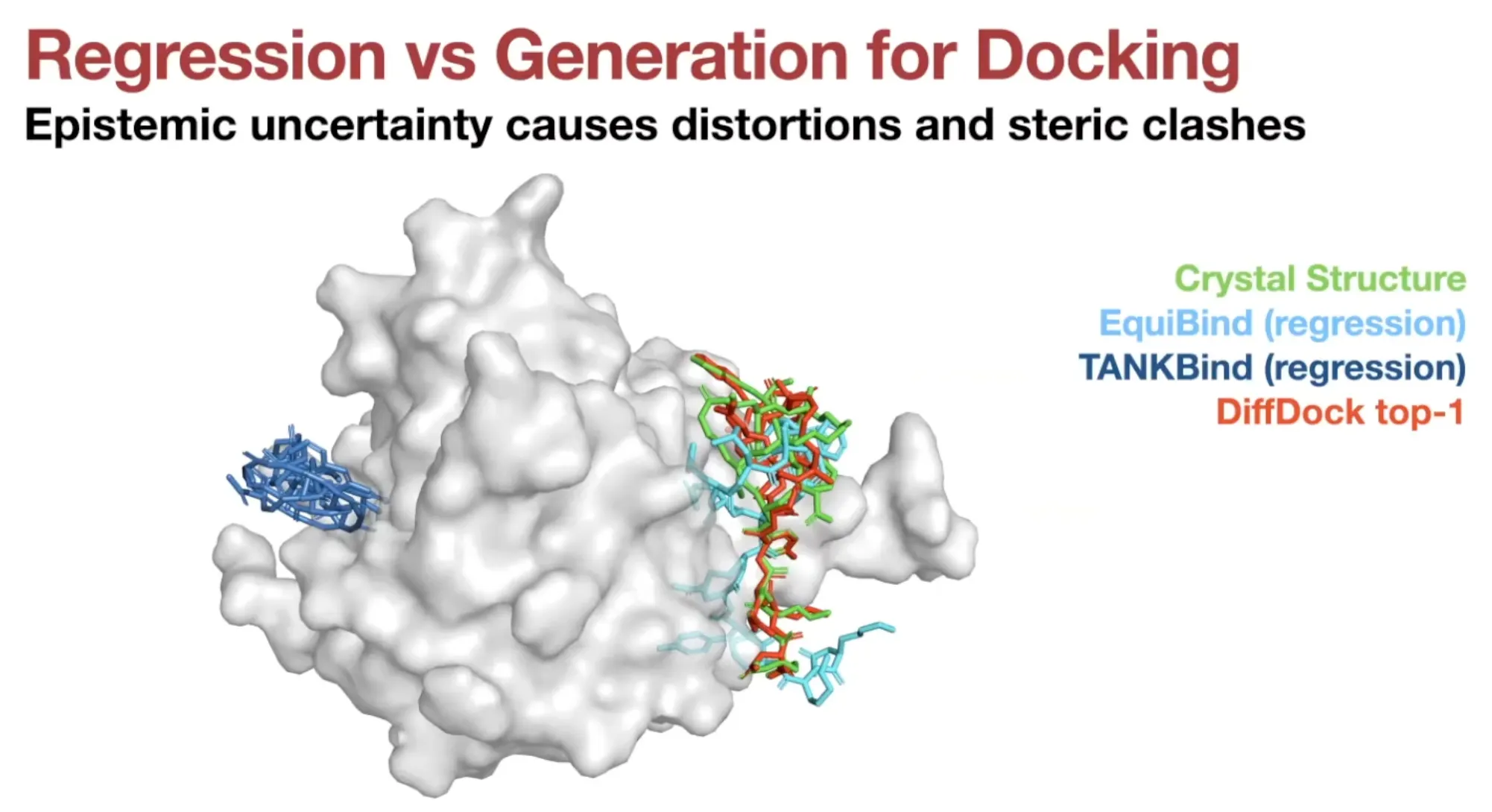
•
Much less steric clashes for generative models
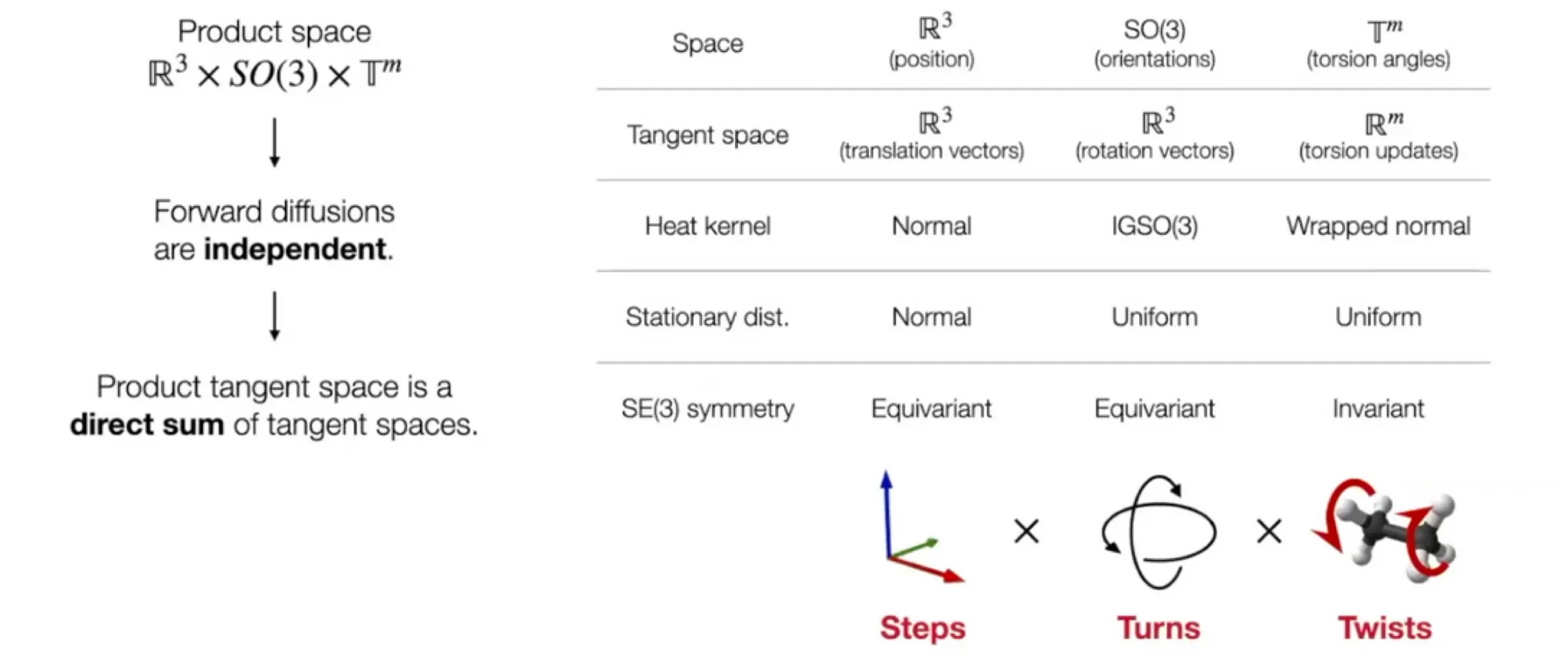
Diffusion Model
DiffDock Overview
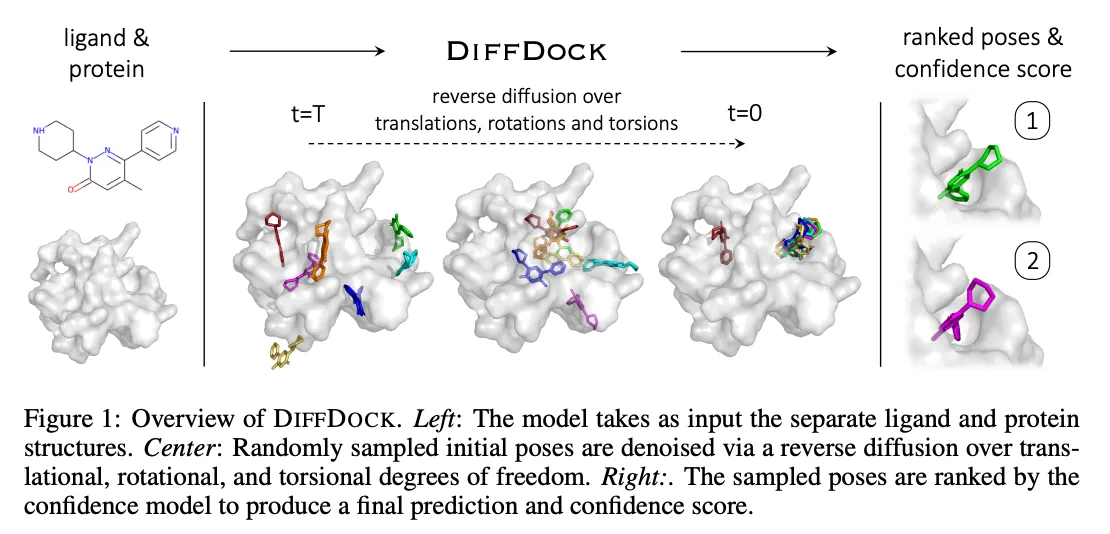
•
Two-step approach
◦
Score model: Reverse diffusion over translation, rotation, and torsion
◦
Confidence model: Predict whether or not each ligand pose is compared to ground truth ligand pose
Score model
•
Ligand pose: (: number of atoms)
•
But molecular docking needs far less degrees of freedom.
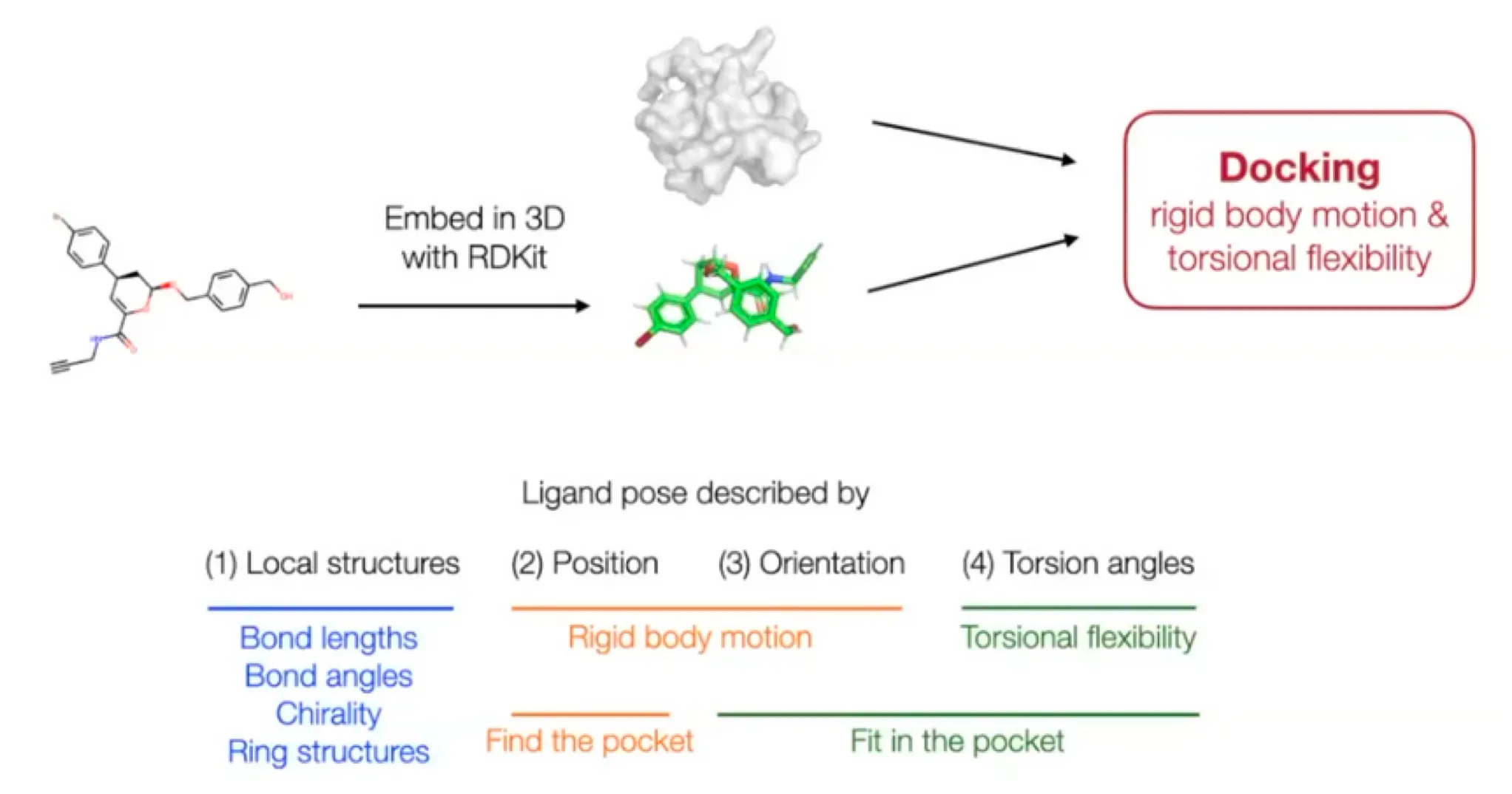
◦
Reduced degree of freedom:
▪
Local structure: Fixed (rigid) after conformer generation with RDKit EmbedMolecule(mol)
•
Bond length, angles, small rings
▪
Position (translation): - 3D vector

▪
Orientation (rotation): - three Euler angle vector
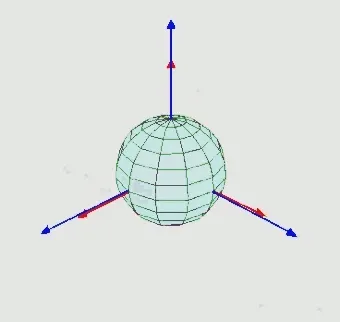
▪
Torsion angles: (: number of rotatable bonds)
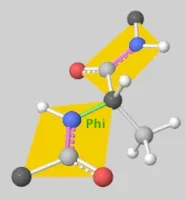
◦
Can perform diffusion on product space
▪
For a given seed conformation , the map is a bijection!
Confidence Model
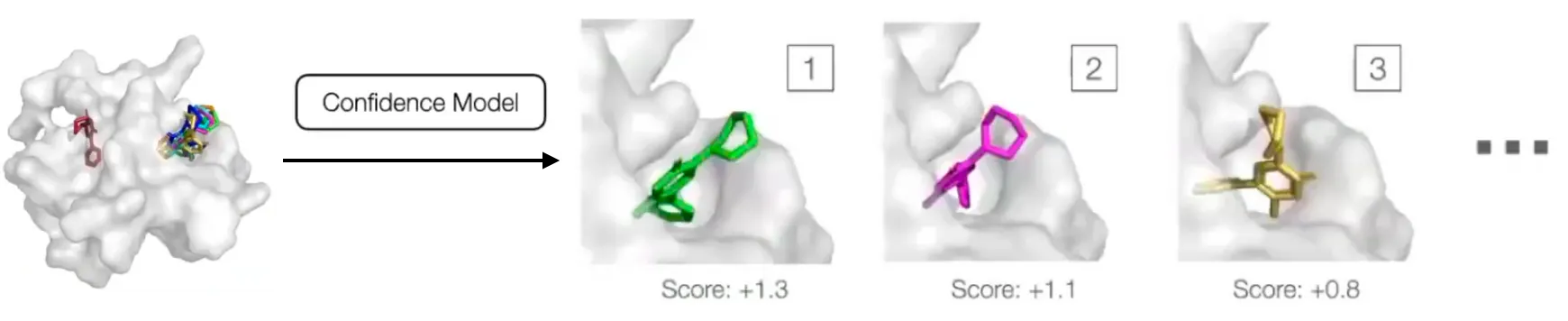
•
Generative model can sample an arbitrary number of poses, but researchers are interested in one or a fixed number of them.
•
Confidence predictions are very useful for downstream tasks.
•
Confidence model
◦
: pose of a ligand
◦
: target protein structure
•
Samples are ranked by score and the score of the best is used as overall confidence score.
•
Training & Inference
◦
Ran the trained diffusion model to obtain a set of candidate poses for every training example and generate binary labels: each pose has RMSD below or not.
◦
Then the confidence model is trained with cross entropy loss to predict the binary label for each pose.
◦
During inference, diffusion model is run to generate poses in parallel, and passed to the confidence model that ranks them based on its confidence that they have RMSD below .
DiffDock Workflow
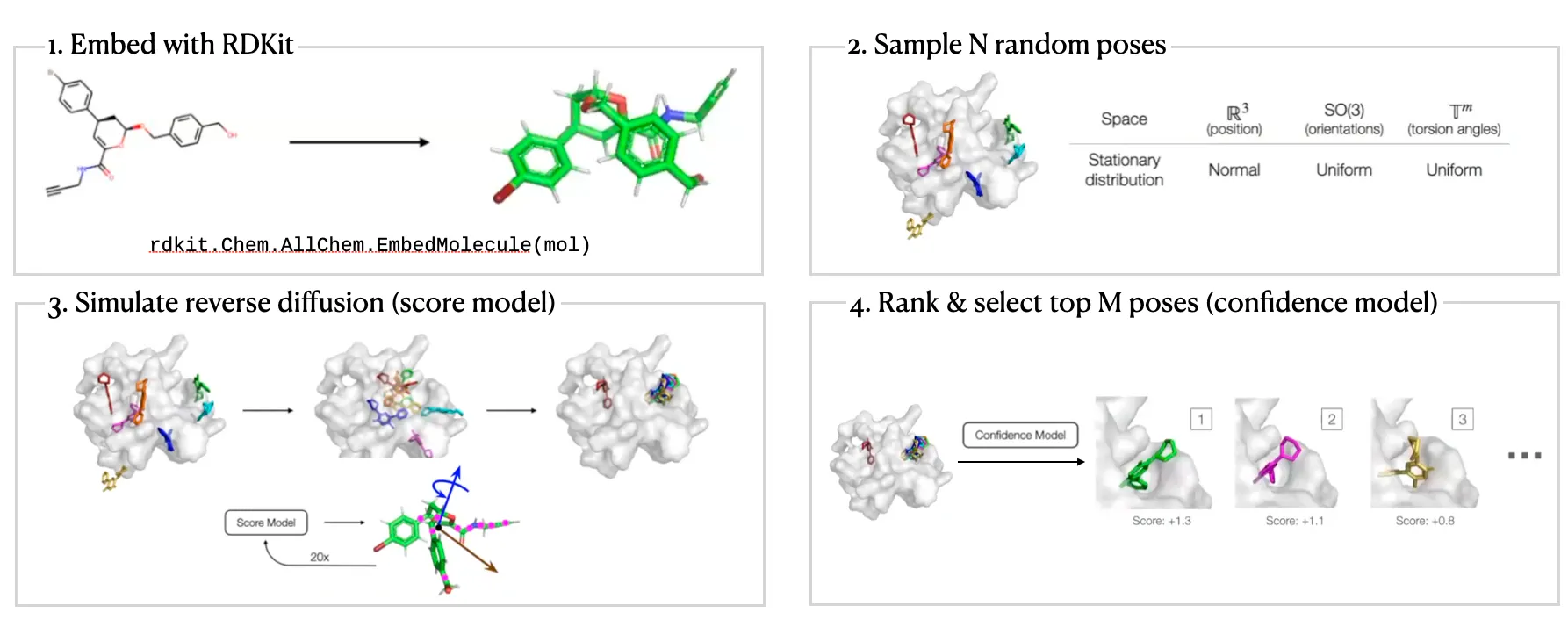
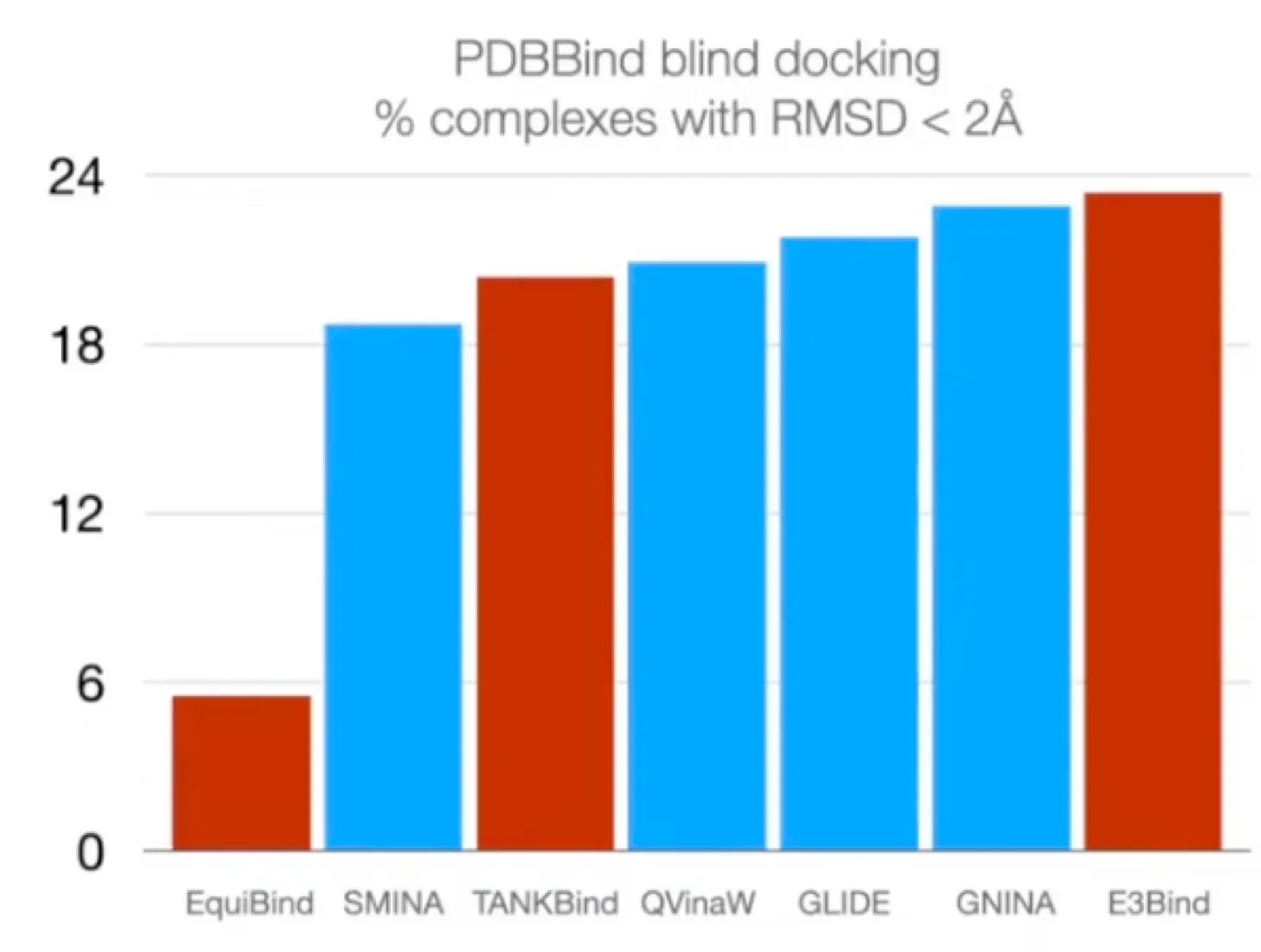
DiffDock Results
Personal opinions
•
It is impressive that the authors formulated molecular docking as a generative problem, conditioned on protein structure.
•
But it is not an end-to-end approach. And there are some discrepancy between the inputs and output of the confidence model. The input is the predicted ligand pose and protein structure , but the output is “whether the RMSE is below 2Å between predicted ligand pose and ground truth ligand pose ”.
•
There are quite a room to improve the performance, but it requires heavy workloads of GPUs.
•
I’m skeptical about the generalizability of this model since there are almost no physics informed inductive bias in the model.
Reference

•
Blog