
Table of Contents
Introduction
If you need some background information about protein folding, refer to  Introduction to Protein Folding
Introduction to Protein Folding 
Introduction to Protein Folding Timeline of AlphaFold
•
2016
◦
AlphaGo beat Lee Sedol
◦
DeepMind hired a handful of biologists
•
2018
◦
CASP13
◦
AlphaFold1
•
2020
◦
CASP14
◦
AlphaFold2
•
2021
◦
AlphaFold-Multimer
•
2023
◦
AlphaFold latest (blog post)
•
2024
◦
AlphaFold3 → This post’s topic!
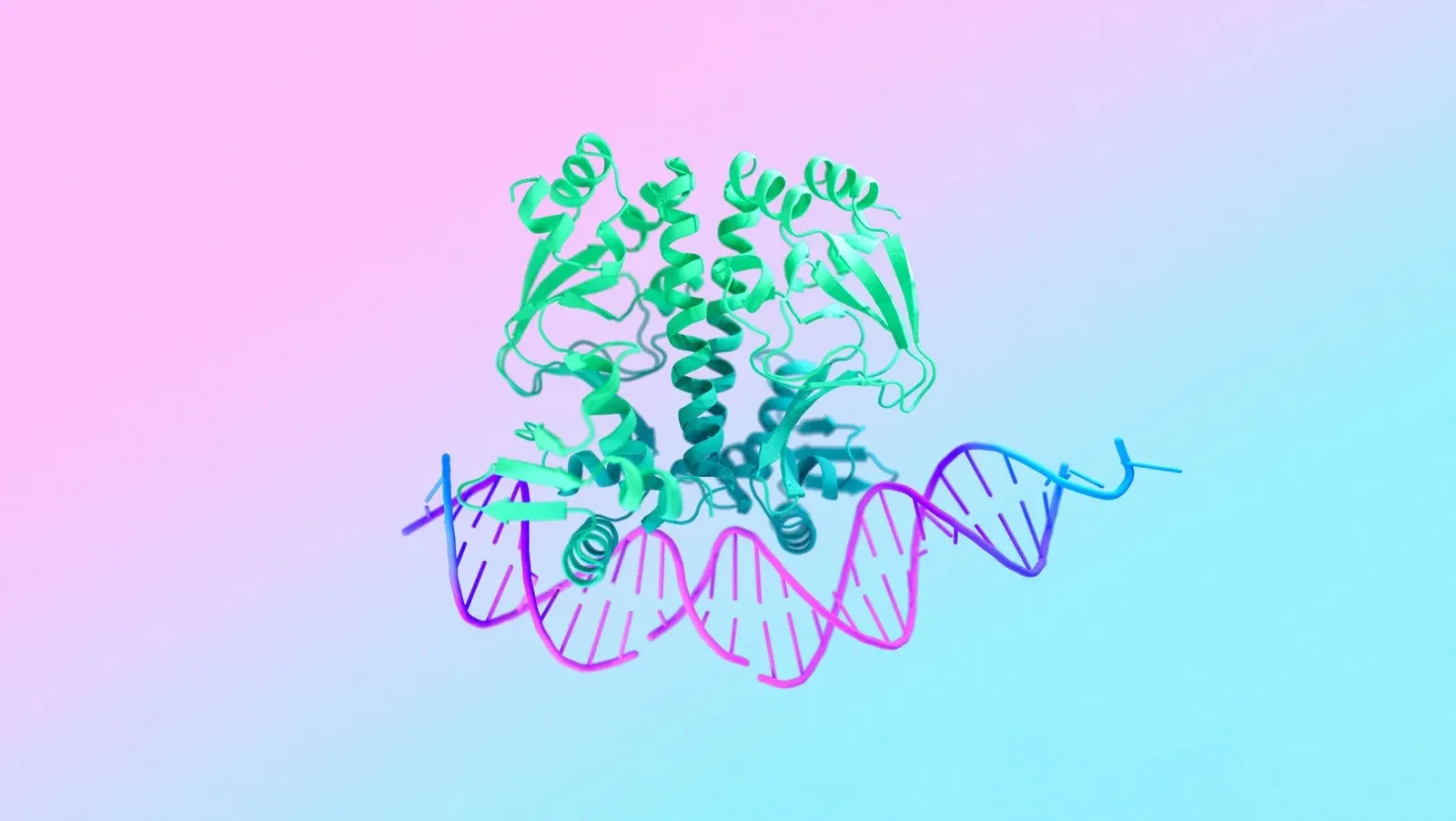
AlphaFold Multimer
Multimeric structure prediction model
Major differences compared to vanilla AF2
•
Multi-chain featurization
◦
asym_id: unique integer per chain
◦
entity_id: unique integer for each set of identical chains
◦
sym_id: unique integer within a set of identical chains
example: A3B2 stoichiometry
•
Multi-chain cropping
◦
Contiguous cropping
◦
Spatial cropping (interface-biased)
•
Symmetry handling
◦
Greedy heuristic approach to deal with multi-chain permutation alignment
•
Loss
◦
FAPE loss cutoff
▪
intra-chain: 10Å (same as vanilla AF2)
▪
inter-chain: 30Å
◦
(new) chain center-of-mass loss term
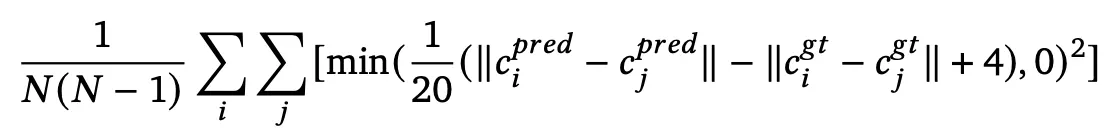
push apart different chains (clamped if the error is -4Å or greater)
Goal: to prevent the model from predicting overlapping chains
◦
(modified) clash loss
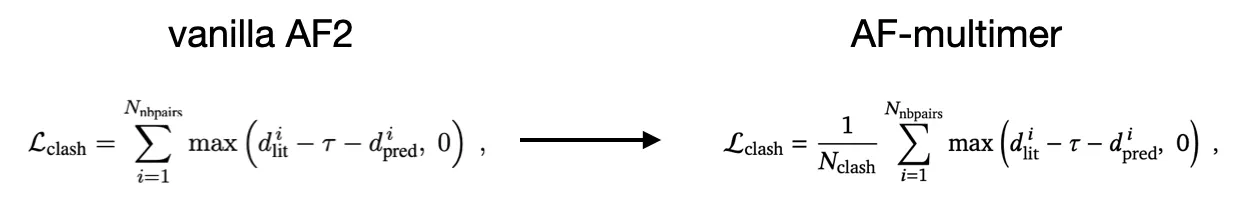
average, rather than sum
Goal: stabilize the loss (since there maybe many clashes if Ncycle is small - due to black hole initialization)
•
Architecture
◦
Template stack
Swapped the order of attention and triangular multiplicative update layers
Changed the aggregation of template embeddings
◦
Evoformer
Moved the outer product mean to the start of the Evoformer block
Results
Structures
Mean DockQ score
Confidence score (interface pTM) vs. DockQ score
AF-multimer predicts better individual chain than AF2?
Discussion
•
Multi-chain version of AF2, with some modifications
AlphaFold3
Almighty AF model for nearly all bio-molecular types
Major differences with AF2-Multimer
Data type & processing
•
Protein (polypeptide) only → Protein, nucleic acids, small molecules, ions, …
•
Amino acid residue frame & angles → tokens & atoms
Atoms are grouped into tokens.
Standard nucleic acid & amino acid: token represents entire nucleotides or residues
Others, each token corresponds to a single heavy atom
Architecture
•
Evoformer → Pairformer
•
Structure module → Diffusion module
Diffusion module conditions on pair and single features.
Initial coordinate
•
Black hole initialization → generated conformer
Activation function
•
ReLU → SwiGLU
Architecture overview
c.f. AF2 architecture

AF3 architecture for inference.
Training & inference scheme is a bit different.
Input processing
Tokenization
amino acid, nucleotide structure
•
Standard amino acid residue: single token
•
Standard nucleotide residue: single token
•
Modified amino acid / nucleotide: tokenized per-atom
•
All ligands: tokenized per-atom
Each token is designated with a token center atom
•
CA for standard amino acid
•
C1’ for standard nucleotide
•
First (and only) atom for others
Features
•
Token features: position, chain identifier, masks, …
•
Reference features: derived from the reference conformer, generated with RDKit ETKDGv3
•
MSA features
•
Template features
•
Bond features: bond information
Template module
Template stack is similar to AF2
MSA module

MSA is used for protein and RNA sequences.
MSA representation is updated somewhat differently with AF2.
In AF2, attention was performed in two axes (row & column). But in AF3, there is no key-query based attention.
The representation is averaged by pair representation based weights.
→ Reduced computation & memory (no attention with MSA rep), impose much information on pair representation
Pairformer

Pairformer module. n: number of tokens, c: number of channels. Each 48 blocks does not share weights.
In AF2, the Evoformer used MSA rep and pair rep. In AF3, the Pairformer use single rep instead of MSA rep.
In Pairformer, there is attention in single rep, but only row-wise attention exists since there is only one row in single rep.
Unlike in AF2, the single rep does not influence the pair representation within a block. i.e. there is no outer product mean in Pairformer.
Diffusion module

Diffusion module. coarse: token, fine: atom. green: input, blue: pair, red: single.
In AF2, the final structure was built by Structure module using IPA. In AF3, it was replaced with standard non-equivariant(!) point-clound diffusion model over all atoms.
•
Two-level architecture: working first on atoms, then tokens, then atoms again.
•
No geometric inductive bias (locality, SE3 invariance, …) involved
Sequence-local atom attention
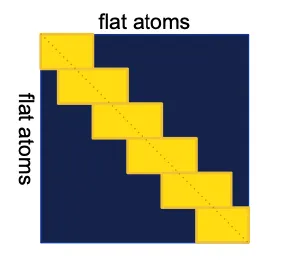
Attention between local sequence level atom neighbors.
This restriction is sub-optimal, but was necessary to keep the memory and compute costs within reasonable bouds.
Diffusion module algorithm
Diffusion Training
•
Efficient training of Diffusion module
Diffusion module is much cheaper than the network trunk (previous modules).
So to improve efficiency, the Diffusion module is trained with larger batch size (48) than the trunk.
•
Loss: weighted aligned MSE loss (upweighting of nucleotide and ligand atoms), bond distance loss (during fine-tuning only), smooth LDDT loss
Training setup

Training setup (distogram head omitted) starting from the end of the network trunk. blue line: abstract activation, yellow: ground truth, green: predicted data
Diffusion “rollout” and confidence module
In AF2, confidence module was trained with the output of the Structure module.
But in AF3, this is not applicable since only a single step of the diffusion is trained (instead of the full structure generation).
Instead, the diffusion module is ran in inference mode (with much larger step size) to produce final structure and this is fed into the confidence module.
Loss
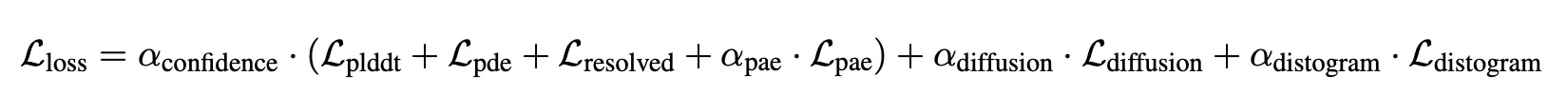
is 1 for final fine-tuning, 0 for others.
Engineering
training protocols